
Abstract
The electrochemical properties of La0.6Sr0.4Co0.2Fe0.8O3-δ-based cathodes are studied as model electrodes for proton ceramic fuel cells. The electrochemical performance of symmetric cells with porous cathodes (La0.6Sr0.4Co0.2Fe0.8O3-δ, La0.6Sr0.4Co0.2Fe0.8O3-δ–BaCe0.9Y0.1O3-δ, and La0.6Sr0.4Co0.2Fe0.8O3-δ–BaZr0.8Y0.2O3-δ), investigated as a function of oxygen and water partial pressures, follows the order La0.6Sr0.4Co0.2Fe0.8O3-δ–BaCe0.9Y0.1O3-δ ≥ La0.6Sr0.4Co0.2Fe0.8O3-δ >> La0.6Sr0.4Co0.2Fe0.8O3-δ–BaZr0.8Y0.2O3-δ. The results indicate that the cathode performance of La0.6Sr0.4Co0.2Fe0.8O3-δ–BaCe0.9Y0.1O3-δ is enhanced mainly due to the extension of the effective triple phase boundary, whereas that of La0.6Sr0.4Co0.2Fe0.8O3-δ–BaZr0.8Y0.2O3-δ is lowered due to the poor proton conductivity along the percolated BaZr0.8Y0.2O3-δ particles. From the observed oxygen partial pressure dependence, the rate-determining step of the above cathode polarization reaction is principally ascribed to the oxygen reduction reaction.

Schematics of the cathode reaction mechanism at the surface of the LSCF, LSCF-BCY, and LSCFBZY cathodes
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Proton ceramic fuel cells (PCFCs) have received much attention because of their advantages over conventional oxide-ion-conducting solid oxide fuel cells (SOFCs). These essential advantages include (i) high fuel utilization and efficiency, since protons react directly with oxygen at the cathode side to form water and prevent fuel dilution during operation; and (ii) operational temperatures of less than 600 °C, which allow the use of less expensive components and increase cell lifetimes [1–4]. The performance of PCFCs is mainly limited by cathode performance, because mixed proton/electron conductivity and high oxygen reduction reaction (ORR) activity are required for PCFC cathodes. It is generally thought that cathode polarization in a PCFC occurs over a series of successive steps: (i) O2 gas diffusion, (ii) dissociative adsorption and surface diffusion of oxygen followed by charge transfer, (iii) H+ migration to the triple phase boundary from the electrolyte, and (iv) the formation and desorption of H2O [5].
Significant effort has been devoted to develop suitable cathodes for PCFCs. Recently, Ba-containing perovskite oxides (PrBaCo2O5+δ and Ba0.5Sr0.5Co0.8Fe0.2O3-δ) [6–9] and the Ruddlesden-Popper oxides (Pr2-xSrxNiO4+δ) [9–11] were reported as promising candidates due to their excellent electrochemical performance. However, the use of these oxides is limited: the basic Ba in the PrBaCo2O5+δ and Ba0.5Sr0.5Co0.8Fe0.2O3-δ can easily react with CO2 in ambient air, leading to the formation of BaCO3 [9], and secondary phases have been observed for Pr2-xBaxNiO4+δ as a result of its reaction with the electrolyte [10]. In another promising approach, it has been demonstrated that introducing a proton-conducting phase into the mixed oxide/electron-conducting phase (composite cathode) can enhance the performance of PCFCs [12–18]. However, because of the low firing temperature of the composite cathode (1000–1200 °C), its proton conducting phase might not be sufficiently sintered to establish proton conducting pathways [12–18]. It is evident that a significant performance enhancement of the PCFC cathode could be obtained upon adding a proton conducting phase (BaZr0.5Pr0.3Y0.2O3-δ, BaCe0.9Yb0.1O3–δ, La5.5WO12−δ, BaZr0.1Ce0.7Y0.1Yb0.1O3 – δ, and BaCe0.8Sm0.2O3 – δ) or even an oxide conducting phase (Sm0.2Ce0.8O2−δ) into the mixed oxide/electron-conducting phase (LSCF, La0.8Sr0.2MnO3−δ, and Nd1.95NiO4+δ). The enhancement would be mainly associated with the extension of the cathode/electrolyte interface. However, a better understanding of composite cathodes is still needed. Using state-of-the-art PCFC materials, this work focuses on elucidating the role of a proton-conducting phase (BaZr0.8Y0.2O3-δ and BaCe0.9Y0.1O3-δ) in La0.6Sr0.4Co0.2Fe0.8O3-δ cathodes in the presence of a BaZr0.8Y0.2O3-δ electrolyte. A systematic electrochemical study is performed to investigate the effects of the oxygen and water partial pressures on the system and elucidate the rate-limiting step at the PCFC cathode.
Experimental
La0.6Sr0.4Co0.2Fe0.8O3-δ (LSCF) powder, prepared by the co-precipitation method, was purchased from Seimi Chemical Co., Ltd. BaZr0.8Y0.2O3-δ (BZY) and BaCe0.9Y0.1O3-δ (BCY) were prepared by conventional solid-state reactions. Stoichiometric amounts of BaCO3 (≥99 %, Sigma-Aldrich), ZrO2 (99 %, Junsei), CeO2 (99 %, Junsei), and Y2O3 (99.99 %, Aldrich) were ball-milled in ethanol for 48 h and dried at 120 °C for 12 h. The respective mixed powders were calcined for 5 h at 1000 °C and 15 h at 1200 °C with intermediate regrinding to obtain pure product phases [19, 20].
For the electrochemical measurements, symmetrical cells were prepared. To obtain dense BZY electrolyte pellets, 2 wt% NiO was added as a sintering aid, as described in the literature [21]. Cathodes were brush-printed (0.39 cm2) on both sides of the electrolyte pellets using cathode slurries consisting of LSCF, LSCF–BCY (70:30 vol%), or LSCF–BZY (70:30 vol%) in a commercial binder (Heraeus, V-006), and then treated at 1000 °C for 5 h in O2 (99.999 %). The sintering of the electrolyte and firing of the cathode were conducted in O2 to prevent reactions with CO2 and H2O in ambient air. The microstructure of the electrodes was examined using a scanning electron microscope (SEM, Hitachi S-4700).
Electrochemical impedance spectroscopy (EIS) measurements were conducted using an Autolab PGSTAT302N (Metrohm) with an oscillation voltage of 50 mV under the open circuit voltage condition in the frequency range 106–10−2 Hz at 600 °C. To investigate the reaction kinetics at the cathode, the polarization resistance was measured at different partial pressures of water (pH2O) and oxygen (pO2). Different pO2 levels were applied by supplying O2, air, or Ar (100 mL‧min−1) into the test chamber. The pO2 values of the dry gases (O2, air, and Ar) were measured by an in-house yttria-stabilized zirconia solid electrolyte sensor operating at 850 °C. The pH2O of the gas was controlled at 3, 5, 10, or 15 % by bubbling the gas through a water flask at 24, 33, 46, or 54 °C, respectively, in a thermostatic bath (CPT, DWB5). The deviations for both pO2 and pH2O were within 1 % of the measured values. Impedance spectra were fitted using ZView software (Scribner Associates, Inc.) and converted to distribution-of-relaxation-time (DRT) plots via MATLAB software (Mathworks), as described in the literature [22–25]. The distribution of the relaxation times (g (τ)) can be written by the following integral equation:
where Z pol is the complex impedance, R pol is the total polarization resistance of the real part of impedance, j is a complex unit, ω is the angular frequency (ω = 2πf), τ is the relaxation time (τ = 1/2πf), and f is the relaxation frequency.
Results and Discussion
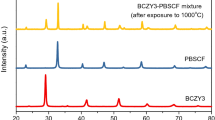
Figure 1 shows the X-ray diffraction patterns of the LSCF, LSCF-BCY, and LSCF-BZY electrodes on the BZY electrolyte after firing at 1100 °C. All the peaks can be indexed to rhombohedral LSCF, orthorhombic BCY, and cubic BZY phases. No impurity phases are observed from the X-ray diffraction data. Figure 2 shows the surfaces and cross-sectional morphologies of the LSCF, LSCF-BCY, and LSCF-BZY electrodes. Because of their similar microstructures and thicknesses (in the range 4–5 μm), the electrode microstructures have negligible effect on the electrochemical performance for this study.

X-ray diffraction patterns of LSCF, LSCF-BCY, and LSCF-BZY electrodes on BZY electrolyte after firing

Surface and cross-sectional SEM images of the LSCF, LSCF-BCY, and LSCF-BZY cathodes
Figure 3a presents the typical EIS spectra of symmetric cells based on the BZY electrolyte with LSCF, LSCF–BCY, and LSCF–BZY cathodes, recorded at 600 °C in wet air (0.03 bar H2O). It has been demonstrated that the proton transport properties of BZY are not dramatically influenced by the addition of 2 wt% NiO as a sintering aid [21]. The total ion transport number of BZY with 2 wt% NiO is higher than 0.92, affording a total ion conductivity of 3.1 × 10−3 S‧cm−1 under wet Ar at 600 °C. Since the partial short circuiting of the electrolyte by 2 wt% NiO is not apparent, the resistance of the electrolyte is subtracted for the following discussion to simplify the presentation. The electrochemical performance of the cathodes follows the order LSCF–BCY ≥ LSCF >> LSCF–BZY. The performance enhancement of LSCF–BCY compared to LSCF can be ascribed mainly to the introduction of proton-conducting percolation pathways from the addition of 30 vol% BCY in the cathode, since it has been generally observed that the percolation threshold of a ceramic composite cathode for a SOFC is around 30 vol% [26]. In contrast, the performance of LSCF–BZY is significantly lower than that of LSCF and is limited by a low frequency process. Despite the introduction of the proton-conducting BZY, an increase in cathodic polarization resistance is observed due to (i) the decrease in available sites for the ORR, (ii) the lowering of electronic conductivity, and (iii) the limited proton conductivity along the BZY particle percolation pathways throughout the LSCF–BZY cathode. The decreases in ORR sites and electronic conductivity do not significantly contribute to the increase in the polarization resistance of LSCF–BZY, because the LSCF–BCY cathode exhibits better cathodic polarization than LSCF. The decrease in the LSCF–BZY cathode performance is mainly a consequence of the sluggish proton conductivity of the BZY particle network; the grain boundary conductivity of BZY is substantially lower (by more than two orders of magnitude at 600 °C) than that of BCY, and BZY is also refractory toward the formation of proton conducting pathways [19, 20]. It can be reasonably concluded that the addition of BZY reduces the effective cathode reaction sites, resulting in the inferior performance of LSCF–BZY. This observation proves that the introduction of a proton-conducting phase in the PCFC cathode is not a preferred way to enhance cathode performance unless the introduced phase can provide sufficient proton conductivity without losing both oxygen reduction activity and electronic conductivity.

a EIS and b DRT plots of the LSCF, LSCF-BCY, and LSCF-BZY cathodes in a symmetric cell at 600 °C under air containing 0.03 bar H2O
To understand the underlying electrochemical mechanism from the EIS data, the DRT transformation was applied to extract the sub-processes which contribute to the overall cathode polarization. The resultant DRT plots from the EIS data are shown in Fig. 3b. The DRT data for the LSCF and LSCF–BCY cathodes confirm that the cathodic process consists of two sub-processes (P1 and P2), whereas the LSCF–BZY cathode exhibits three sub-processes (P1, P2, and P3). Sub-process P1 is attributed to the formation of adsorbed atomic oxygen species by charge transfer, which can directly react with protons to form water at the triple phase boundary, whereas sub-process P2 is associated with the dissociative adsorption of oxygen over the entire cathode surface [5–12]. The P3 sub-process is only apparent for the LSCF–BZY cathode due to its low number of surface reaction sites and poor proton conductivity. Similar results have been reported for La0.8Sr0.2MnO3-δ and LSCF-based electrodes [14, 27]. The LSCF–BZY cathode was not investigated further since the P3 sub-process is beyond the EIS measurement frequency range. On the basis of the DRT analysis, all the EIS data from the LSCF and LSCF–BCY cathodes were fitted using an equivalent circuit with R(R HF Q HF)(R LF Q LF), where R is the resistance of the BZY electrolyte, and the two resistance components (R HF and R LF) and constant phase elements (Q HF and Q LF) in the high (HF) and low frequency (LF) ranges correspond to the cathode polarization process. From the fitting results, the equivalent capacitance C eq of each process was calculated as described in the literature [8–11] using the following equation:
where n shows the deviation of the constant phase element from the ideal capacitance. The total polarization resistance (R p ) is obtained as the sum of R HF and R LF. The EIS fitting parameters are given in Table 1. Schematic illustrations of the electrode reactions of the LSCF, LSCF–BCY, and LSCF–BZY cathodes are shown in Fig. 4.

Schematic representation of the cathode reaction mechanism proposed for the LSCF, LSCF-BCY, and LSCF-BZY cathodes
Figure 5a, b show the EIS spectra of the LSCF and LSCF–BCY cathodes as a function of pH2O. The decreases in R HF and R LF for LSCF–BCY suggest that the extension of the triple phase boundary by the addition of the proton-conducting BCY facilitates the cathode polarization reaction. The overall cathode reactions of LSCF and LSCF–BCY are limited by the HF process, which is also presented in the DRT data. As shown in Fig. 5c, the calculated values of the HF and LF capacitance are in the ranges 10−5–10−4 F and 10−1–101 F, respectively, showing a weak dependence on pH2O, in good agreement with the values in the literature [8–11]. On the other hand, the obtained R HF and R LF increase concomitantly with pH2O (see Fig. 5d). These results are mainly due to the suppression of water formation and gas evolution from the cathode, representing a shift to the left in the cathode reaction equilibrium, \( 2{\mathrm{O}\mathrm{H}}_{\mathrm{O}}^{\bullet }+\frac{1}{2}{\mathrm{O}}_2+2{\mathrm{e}}^{-}\leftrightarrow {\mathrm{H}}_2\mathrm{O}+2{\mathrm{O}}_{\mathrm{O}}^{\times } \) [9–11]. The data obtained in this study are in good agreement with previously reported composite cathodes (LSCF–BaCe0.9Yb0.1O3–δ [13], La0.8Sr0.2MnO3−δ–La5.5WO12−δ [14], Nd1.95NiO4+δ–BaZr0.1Ce0.7Y0.1Yb0.1O3 − δ [16], and LSCF–BaZr0.5Pr0.3Y0.2O3-δ [18]). Figure 6 shows the long-term performance of the LSCF-BCY composite cathode at 600 °C under 15 % H2O/air atmosphere. The overall polarization resistance of the LSCF-BCY cathode exhibits a stable value without deterioration for more than 50 h. The post microstructure analysis of the LSCF-BCY cathode by means of SEM did not reveal any delamination of the composite cathode.

EIS spectra under a pH2O gradient from 0.03 to 0.15 bar at 600 °C under air for the a LSCF and b LSCF-BCY cathodes. The equivalent fitting results are shown as lines. Variation of calculated c capacitance (C eq) and d area-specific resistance values (R p, R HF, and R LF) as a function of pH2O for the LSCF (★, ☆) and LSCF-BCY (▲, △) cathodes

Long-term performance of LSCF-BCY composite cathode at 600 °C under 15 % H2O/air atmosphere for 52 h
Figure 7a illustrates the EIS plot of the LSCF–BCY cathode as a function of pO2. The cathode impedance is dramatically increased with decreasing pO2 at constant pH2O. The resulting polarization resistance values are shown in Fig. 6b–d. The total polarization resistance shows evidence for a change-over of the limiting process from HF to LF below a pO2 of ∼10−2 bar. Possible pathways for the ORR [28] at the PCFC cathode are (i) oxygen adsorption (O2 ↔ O2,ad), (ii) dissociative oxygen adsorption (O2 + 2e′ ↔ 2O −ad ), and (iii) charge transfer (O −ad + e′ ↔ O 2 −TPB ), and (iv) an oxygen incorporation reaction (O −ad + e′ + V • •O ↔ O xO ), where e′ and V • •O are the electron and oxygen vacancy at the cathode surface, respectively. The pO2 dependence of the cathode polarization resistance as a function of pO2 is summarized in Table 2. The observed average pO2 dependence values for the elementary steps are −0.22 and −0.53 for R HF and R LF, respectively. The −0.25 power dependence can be assigned to the charge transport of the adsorbed oxygen species at the triple phase boundary (O −ad + e′ ↔ O 2 −TPB ) and/or oxygen incorporation into the oxide lattice (O −ad + e′ + V • •O ↔ O xO ), while the −0.5 power dependence is associated with the dissociative adsorption of oxygen (O2 + 2e′ ↔ 2O −ad ) [13, 16, 17, 29]. Clearly, the ORR at the electrolyte/cathode interface and/or cathode surface is crucial in order for H+ from the electrolyte to react to form water, which is then released to the gas phase. An important corollary of this study is that, even though protons are the major charge carriers in the PCFC electrolyte, the ORR in the LSCF-based PCFC cathode is the rate-limiting step and is independent of proton transport.

a EIS spectra of the LSCF-BCY cathode under an pO2 gradient from 1 to 10−4 bar containing 0.03 bar H2O at 600 °C. The equivalent fitting result is shown as lines. Variations of the calculated b area-specific overall polarization resistance (R p ), c high frequency polarization resistance (R HF), and d low frequency polarization resistance (R LF) as a function of pO2 for the LSCF (★, ☆) and LSCF-BCY (▲, △) cathodes
Conclusions
In this study, we examined the electrochemical properties of LSCF-based PCFC cathodes. Our observations are summarized as follows:
-
1.
The electrochemical performance of the cathode increases with the extension of the effective triple phase boundary at the electrolyte/cathode interface and cathode surface, which comprises a proton conductor with high grain boundary conductivity.
-
2.
The polarization resistance increases with increasing pH2O due to the suppression of water formation.
-
3.
The observed pO2 dependence of the polarization resistance suggests that the rate-determining step at the cathode is the ORR, rather than proton transport.
References
H. Iwahara, Solid State Ionics 77, 289 (1995)
N. Bonanos, Solid State Ionics 79, 161 (1995)
L. Bi, E. Traversa, J. Mater. Res. 29, 1 (2013)
L. Bi, S. Boulfrad, E. Traversa, Chem. Soc. Rev. 43, 8255 (2014)
F. He, T. Wu, R. Peng, C. Xia, J. Power Sources 194, 263 (2009)
Y. Lin, R. Ran, Y. Zheng, Z. Shao, W. Jin, N. Xu, J. Ahn, J. Power Sources 180, 15 (2008)
Y. Lin, R. Ran, C. Zhang, R. Cai, Z. Shao, J. Phys. Chem. A 114, 3764 (2010)
J. Dailly, S. Fourcade, A. Largeteau, F. Mauvy, J.C. Grenier, M. Marrony, Electrochim. Acta 55, 5847 (2010)
A. Grimaud, F. Mauvy, J.M. Bassat, S. Fourcade, L. Rocheron, M. Marrony, J.C. Grenier, J. Electrochem. Soc. 159, B683 (2012)
P. Batocchi, F. Mauvy, S. Fourcade, M. Parco, Electrochim. Acta 145, 1 (2014)
A. Grimaud, F. Mauvy, J.M. Bassat, S. Fourcade, M. Marrony, J.C. Grenier, J. Mater. Chem. 22, 16017 (2012)
T. Wu, Y. Zhao, R. Peng, C. Xia, Electrochim. Acta 54, 4888 (2009)
E. Fabbri, S. Licoccia, E. Traversa, E.D. Wachsman, Fuel Cells 9, 128 (2009)
C. Solís, L. Navarrete, S. Roitsch, J.M. Serra, J. Mater. Chem. 22, 16051 (2012)
G. Taillades, P. Pers, P. Batocchi, M. Taillades, ECS Trans. 57, 1289 (2013)
C. Yang, X. Zhang, H. Zhao, Y. Shen, Z. Du, C. Zhang, Int. J. Hydrog. Energy 40, 2800 (2015)
L. Zhao, B. He, J. Gu, F. Liu, X. Chu, C. Xia, Int. J. Hydrog. Energy 37, 548 (2012)
E. Fabbri, L. Bi, D. Pergolesi, E. Traversa, Energy Environ. Sci. 4, 4984 (2011)
E. Fabbri, D. Pergolesi, S. Licoccia, E. Traversa, Solid State Ionics 181, 1043 (2010)
G. Chiodelli, L. Malavasi, C. Tealdi, S. Barison, M. Battagliarin, L. Doubova, M. Fabrizio, C. Mortalo, R. Gerbasi, J. Alloys Compd. 470, 477 (2009)
C.-Y. Yoo, D.S. Yun, J.H. Joo, J.H. Yu, J. Alloys Compd. 621, 263 (2015)
A.L. Smirnova, K.R. Ellwood, G.M. Crosbie, J. Electrochem. Soc. 148, A610 (2001)
H. Schichlein, A.C. Müller, M. Voigts, A. Krügel, E. Ivers-Tiffée, J. Appl. Electrochem. 32, 875 (2002)
S. Hershkovitz, S. Baltianski, Y. Tsur, Solid State Ionics 188, 104 (2011)
B.A. Boukamp, Electrochim. Acta 154, 35 (2015)
D. Chen, L. Lu, J. Li, Z. Yu, W. Kong, H. Zhu, J. Power Sources 196, 3178 (2011)
D.S. Yun, J.H. Joo, J.H. Yu, H.C. Yoon, J.-N. Kim, C.-Y. Yoo, J. Power Sources 284, 245 (2015)
C. Yoo, Phase stability and oxygen transport properties of mixed ionic–electronic conducting oxides, Ph.D. Thesis University of Twente (2012), pp. 14–15
C. Zhang, H. Zhao, J. Mater. Chem. 22, 18387 (2012)
Acknowledgments
This work was conducted under the framework of the Research and Development Program of the Korea Institute of Energy Research (KIER) (B6-2456-01). This research was also supported by the C1 Gas Refinery Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (2015M3D3A1A01064928). Jong Hyun Park (Chungnam National University) and Hyejin Yu (Pukyong National University) are gratefully acknowledged for experimental supports and fruitful discussions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Yoo, CY., Yun, D.S., Park, SY. et al. Investigation of Electrochemical Properties of Model Lanthanum Strontium Cobalt Ferrite-Based Cathodes for Proton Ceramic Fuel Cells. Electrocatalysis 7, 280–286 (2016). https://doi.org/10.1007/s12678-016-0306-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12678-016-0306-1