
Abstract
Manganese oxide-modified graphene nanosheet-supported silver nanocatalyst (Ag-MnOx/G) was prepared via two-step chemical and electrochemical deposition. Surface characterization of the prepared Ag-MnOx/G catalyst was performed by X-ray photoelectron spectroscopy, scanning electron microscopy, as well as X-ray fluorescence techniques, and the electrocatalytic activity toward the oxygen reduction reaction (ORR) in alkaline media was studied using cyclic voltammetry and the rotating disk electrode (RDE) method. The onset potential of the ORR of the prepared catalyst material shifted positive about 40 mV, and the half-wave potential 20 mV compared to those of the bulk Ag electrode. After 1000 potential cycles between 0.05 and 1.1 V for accelerated aging tests, high stability of the Ag-MnOx/G catalyst in the ORR was observed with the half-wave potential of the ORR shifting negatively only about 0.04 V. RDE studies displayed unconditional improvement of electrochemical activity and long-term durability for the Ag-MnOx/G composite material.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cutdown of the amount of platinum metal in catalyst content and development of new catalytic materials for clean energy applications is under extensive research attention. Establishment of noncorrosive, platinum-free inexpensive electrocatalyst for oxygen reduction reaction (ORR) is crucial in modern fuel cell development [1, 2]. Intense research activity in last decade yielded considerable amount of high surface area carbon-based composites [3–5]. Among the numerous non-Pt catalyst candidates toward the ORR, such as other precious metals and their alloys [6, 7], metal macrocycles [8, 9], N-containing high-area carbon [10–17], and different transition metal oxides, especially manganese oxides intercalated to carbon-based composites demonstrated itself as promising cathode materials for fuel cells [18–24]. Manganese oxide (MnOx) is one of the most promising alternatives with considerable catalytic activity toward the ORR with up-and-coming advantages, such as wide natural abundance, low cost, and environmental friendliness [25–28]. Surface morphology of MnOx-based catalysts is an important influential factor to their electrochemical properties [29]. Earlier results show that the electrocatalytic activities of MnO2 toward the ORR depend strongly on the crystallographic structures, following an order of β-MnO2<λ-MnO2<γ-MnO2<α-MnO2≈δ-MnO2 [30]. The ORR activity at different manganese oxides follows trend: Mn5O8<Mn3O4<Mn2O3<MnOOH [31, 32].
On the other hand, silver is also a promising catalyst for ORR [33]. Being less expensive than Pt silver has shown relatively high ORR activity and stability in alkaline electrolyte, and now, it is considered as feasible alternative to replace platinum in cathode catalysts for fuel cells [34–37]. According to earlier studies, Ag has been proven relatively stable in alkaline electrolytes at a wide range of temperatures [38–41]. Wu and coworkers obtained 23 times higher mass activity on the Ag−MnOx/C composite than on the Ag/C (46 vs 2 mA/μg) [42]. Silver has shown high electrical conductivities, especially when supported on conductive supports, such as graphene and carbon, yielding enhanced surface area of active sites [43, 44]. Chatenet et al. studied electrochemical behavior of carbon-supported silver catalysts in alkaline electrolytes and compared its electrocatalytic activity with that of Pt/C. Their study confirmed that the ORR mechanism on carbon-supported Ag in alkaline media shows many similarities with that on Pt/C [45]. Fazil et al. tested carbon nanotube (CNT)-supported Ag catalysts in a fuel cell, and 40 wt.% Ag/CNT showed high activity with peak power density of 26.1 mW/cm2 [46]. The influence of metal loading on Ag/C catalyst was studied, and the optimum loading of silver was found to be around 20 wt.%, while further increase in metal loading decreased the catalyst performance [47]. The predominance of the 4-electron ORR pathway on Ag/C is widely recognized; nevertheless, many reports conclude that small amount of hydrogen peroxide could still be produced as an intermediate [35, 41, 48].
The presence of Ag+ ions in MnO2 structure has been demonstrated to offer high activity in CO oxidation [49], and excellent performances in other catalytic processes due to the large surface area and abundant lattice of Ag-MnOx composites [50–53]. Kostowskyj et al. prepared carbon nanotube-supported silver-manganese oxide nanowires with very low loading (1.82 mg nanowires/500 mg CNTs), and they had a comparable activity to that of several bulk catalysts studied in literature [51]. Although excellent catalytic properties were found in different applications, the electrochemical properties of Ag-MnOx composites have seldom been studied in detail.
This paper reports a systematic study on the electrochemical properties of Ag-MnOx/graphene-based nanostructure as low-cost catalysts for oxygen reduction reaction in alkaline media. Ag-MnOx/graphene composites have been prepared using MnSO4 and KMnO4 as the precursors, and the catalytic activity and stability of the prepared catalysts toward the ORR in 0.1 M KOH solution have been studied and compared with the bulk Ag and Pt electrodes in detail.
Experimetal
Preparation of Ag-MnOx/Graphene Electrocatalyst
The graphene nanoplatelets (SBET = 750 m2/g, an oxygen content of <2 wt.%, and a carbon content of >98 wt.%) were bought from Strem Chemicals, Inc. All the other chemicals were of analytical reagent grade and used as received without any further treatment. All the aqueous solutions and suspensions used were prepared using Millipore ultrapure water (18.2 MΩ cm).
The manganese oxide nanoparticles were chemically deposited onto graphene surface as follows [54–57]: 0.12 g of graphene was mixed with a 2 mL of an aqueous solution containing 10 mM MnSO4 (Aldrich). The suspension was maintained at 80 °C for 20 min under stirring, in order to allow impregnation of the graphene surface by manganese sulfate.
A 4-mL aqueous solution, containing 33 mmol of KMnO4 (Merck) preheated to 80 °C, was added to the suspension during vigorous stirring. Suspension was stirred for 15 min at 80 °C and then filtered and washed 3 times with water. Product was dried at 100 °C for 4 h.
Corresponding mass ratios in the obtained MnOx/G material were 60 mg of graphene to 40 mg of Mn (60 % graphene and 40 % Mn). MnSO4 is oxidized by the permanganate in the presence of graphene according to the following chemical reaction [54]:
For preparation of catalyst ink 10 mg of MnOx/G was suspended in 4 mL of 0.5 wt.% Nafion (Aldrich) solution in ethanol by sonication for 15 min. Five microliters of this suspension was transferred to the polished glassy carbon (GC) electrode surface (A = 0.196 cm2) by pipetting and dried for 5 min at 60 °C. Ag was electrodeposited onto the MnOx/G-modified GC electrode surface from 1 mM AgNO3 solution containing 0.1 M KNO3. The electrodeposition experiments were carried out in a three-electrode cell with MnOx/G-modified GC as working electrode, Pt wire as counter electrode, and saturated calomel electrode (SCE) as a reference electrode. The potential of −0.5 V versus SCE was applied for 30 s.
Surface Characterization
For surface morphology studies, catalyst samples were prepared by modification of GC electrode with MnOx/G powder suspension in 2-propanol (10 mg in 4 mL) followed by subsequent Ag electrodeposition. The appearance of electrochemically deposited Ag particles on MnOx/G was confirmed by scanning electron microscopy (SEM). Energy-dispersive X-ray spectroscopy (EDX) was used to quantitatively identify distribution of silver and manganese oxide particles on graphene support surface. X-ray photoelectron spectroscopy (XPS) and X-ray fluorescence (XRF) techniques were applied to gain more information about the catalyst surface composition. The XPS measurements were performed with a SCIENTA SES-100 spectrometer using non-monochromatized Al Kα X-ray source (1486.6 eV), a takeoff angle of 90°, and a source power of 400 W. The pressure in the analysis chamber was less than 10−9 Torr. For collecting the survey spectra, the following parameters were used: energy range 800–0 eV, pass energy 200 eV, and step size 0.5 eV. In specific regions, high-resolution scans were performed with the pass energy of 200 eV and the 0.1-eV steps. The nominal Mn2O3 and Ag film thicknesses were measured by X-ray fluorescence spectrometer Rigaku ZSX 400 and program ZSX Version 5.55.
Electrochemical Measurements
The potential was applied with an Autolab potentiostat/galvanostat PGSTAT30 (EcoChemie B.V., The Netherlands), and the electrochemical experiments were controlled with the General Purpose Electrochemical System (GPES) software. Cyclic voltammetry (CV) tests were performed in a three-electrode glass cell, where reversible hydrogen electrode (RHE) was used as a reference electrode and a Pt foil as a counter electrode. The GC disk electrode coated with catalyst ink served as working electrode. GC disks (GC-20SS, Tokai Carbon) were pressed into a Teflon holder and were polished to a mirror finish with 1- and 0.3-μm alumina slurries (Buehler). After polishing, the electrodes were sonicated in isopropanol and Milli-Q water for 5 min.
Supporting electrolyte comprised 0.1 M aqueous KOH (p.a. quality, Merck) solution, which was saturated with Ar (99.999 %, AGA) or O2 gas (99.999 %, AGA). Rotating disk electrode (RDE) measurements were carried out at various electrode rotation rates (ω) using RDE setup with CTV101 speed control unit and EDI101 rotator (Radiometer).
The RDE results of O2 reduction were compared with those obtained with the bulk Ag and Pt electrodes under the same measurement conditions. The potential scan rate (ν) used for the oxygen reduction measurements was 10 mV/s, and for stability testing 20 mV/s.
Results and Discussion
Surface Characterization of Ag-MnOx/G Catalyst
SEM images of the unmodified graphene, MnOx/G support, and Ag-MnOx/G catalyst deposited on GC are shown in Fig. 1a, b, c, respectively. Aggregates of submicron graphene nanoplatelets with a diameter of <2 μm and a thickness of a few nanometers are clearly seen in Fig. 1a. The surface morphology of the MnOx/G support material is quite similar to that for pure graphene nanosheets; however, after local magnification, thin layer of metal oxide distributed all over the graphene support can be distinguished. Figure 1c shows that the Ag particles for the Ag-MnOx/G catalyst are the aggregates of the nanoparticles, which are about 12 nm. Meanwhile, the electrodeposited Ag nanoparticles are not distributed uniformly, and some uncovered MnOx/G support surface sites are present. The EDX analysis revealed the atomic composition of the catalyst material, and 4.6 wt.% of silver and 9.4 wt.% of manganese were obtained.

SEM images of a pure graphene nanosheets, b MnOx/G, and c Ag-MnOx/G-modified GC electrodes
In order to further investigate the valence states of Mn and Ag on the surface of the prepared catalyst material, the XPS experiments were performed on the Ag-MnOx/G, and the results are shown in Fig. 2. The binding energies (BE) of Mn2p3/2 and Mn2p1/2 are 641.9 and 653.2 eV, respectively, which shows that manganese consists of a mixture of Mn3+ and Mn4+ (inset to Fig. 2). Although the average oxidation states of manganese cannot be determined from analyses of Mn2p binding energies [58], still, the XPS spectra of O1s has three separate peaks at 528.8, 530.6, and 532.5 eV. These BE values can be attributed to three different types of oxygen bonding, where 528.8 eV corresponds to the binding energy of oxygen bonded to manganese, 530.6 eV corresponds to activated oxygen adsorbed on the surface of the catalyst, and 532.5 eV corresponds to adsorbed water and −OH groups [59]. The Ag3d transition peaks were also evident at BE values of 368 eV for Ag3d5/2 and 374 eV for Ag3d3/2 (inset of Fig. 2), which are typical for Ag+ and Agq+ clusters [59, 60].

XPS spectra for Ag-MnOx/G-modified GC electrodes (insets show the core-level spectra in the Mn2p and Ag3d regions)
Complimentary XRF measurements revealed that the nominal Mn2O3 layer thickness was 9–10 nm, and the thickness of Ag was 5 nm. The Mn/O mass thickness ratios for Ag-MnOx/G samples were 2.32, when theoretical mass thickness Mn/O ratio for Mn2O3 is 2.29.
Cyclic Voltammetry
For the electrochemical characterization of the catalysts, the cyclic voltammetry (CV) curves for the Ag-MnOx/G and MnOx/G modified GC, bulk Ag, and bulk Pt electrodes were recorded with a scan rate of 0.05 V/s in the potential range between 0.05 and 1.4 V in Ar-saturated 0.1 M KOH solution and presented in Fig. 3. For all Ag-containing catalysts, the cathodic silver surface oxide reduction peaks in the potential range between 1.0 and 1.2 V were evident. These cathodic peaks are assigned to the transformation of silver oxides to metallic silver that exist for Ag-MnOx/G and Ag/G-modified GC and bulk Ag electrodes. This peak for Ag-MnOx/G catalyst is relatively weak, which is attributed to the smaller amount of Ag electrodeposited on the MnOx. A pair of redox peaks observed at approximately 1.0 and 0.55 V, respectively, are associated with the formation of MnOOH from MnO2 [61]. For MnOx/G-modified GC electrode, another reduction peak was well defined at approximately 0.7 V. It was previously stated that first, the reduction of O2 to HO2¯ takes place at approximately 0.7 V and the electrochemical transition of HO2¯ to OH¯ takes place at more negative potentials [62].

CV curves of Ag-MnOx/G, MnOx/G-modified GC, and bulk Pt and Ag electrodes in Ar-saturated 0.1 M KOH, ν = 50 mV/s
Rotating Disk Electrode Studies of O2 Reduction
RDE measurements were performed to determine the predominant pathway of oxygen electroreduction. For the RDE experiments, the pure Faradic current under O2 was obtained by subtraction from the background current obtained under argon in the same voltammetry sweep condition. ORR polarization data recorded with the Ag-MnOx/G composite catalyst in 0.1 M KOH at an RDE are shown in Fig. 4a.

a ORR polarization curves for Ag-MnOx/G catalyst in O2-saturated 0.1 M KOH at different electrode rotation rates, b Koutecky–Levich plots of O2 reduction in 0.1 M KOH at different potentials (inset the dependence of n on potential). Data derived from Fig. 4a, c RDE voltammogram comparison for GC, graphene, MnOx/G, Ag/G, bulk Ag, Ag-MnOx/G, bulk Pt in O2-saturated 0.1 M KOH, rotation speed: 1900 rpm, ν = 10 mV/s. d The corresponding Tafel plots of O2 reduction in 0.1 M KOH (derived from c)
RDE results were analyzed using the Koutecky–Levich (K–L) equation [63]:
where j is the measured current density, j k and j d are the kinetic and diffusion-limited current densities, respectively, k is the electrochemical rate constant for O2 reduction, \( {D}_{O_2} \) is the diffusion coefficient of oxygen (1.9 × 10-5 cm2/s) [64], \( {C}_{O_2}^b \) is its concentration in the bulk (1.2 × 10–6 mol/cm3) [64], and v is the kinematic viscosity of the solution (0.01 cm2/s1) [65].
The K–L plots of O2 reduction on Ag-MnOx/G composite are shown in Fig. 4b. The K–L lines are parallel, and the extrapolated lines yield intercepts other than zero indicating that the process of oxygen reduction is under the mixed kinetic–diffusion control in the range of potentials studied. The number of electrons transferred per O2 molecule (n) was calculated from the slope of the K–L lines shown in the inset of Fig. 4b.
The catalytic activity and diffusion current density values for the Ag-MnOx/G composite are much higher than that of Ag/G and MnOx/G composites due to the intrinsic synergy of silver and manganese oxide. The ORR limiting current density at 1900 rpm on Ag-MnOx/G catalyst was approximately 5.51 mA/cm2, which is similar to that of bulk Ag and higher than Ag/G and MnOx/G composites (4.45 and 4.47 mA/cm2, respectively), but slightly lower than that of the bulk Pt electrode (6.15 mA/cm2). The onset potential for Ag-MnOx/G was approximately 0.9 V, which is very close to that for bulk Pt (0.95 V). Two distinct Tafel slopes in two potential regions (0.9>E>0.8 V and 0.8>E>0.7 V) were found for all catalyst under study (Table 1). For Ag-MnOx/G catalyst material, the slope values were −0.057 and −0.122 V per decade at low and high overpotentials, respectively, which indicates that the ORR mechanism is similar to that on platinum. Wu et al. presented similar results for Ag−MnOx/C composites, the Tafel slope values were about −55 and −120 mV/dec at low and high overpotentials, respectively [42]. The comparable Tafel behavior for the electrodes studied was also obtained by Tang et al. indicating that the ORR mechanism is the same, where the one-electron transfer is the rate-determining step at low overpotentials and the two-electron transfer reaction is the rate-determining step at the higher overpotentials [52].
The as-prepared Ag-MnOx/G composite exhibits an onset potential of ~0.9 V and an overall 4-electron transfer involved in the ORR, indicating its potential application as the cathode catalyst for alkaline membrane fuel cells. The superior ORR activity of Ag-MnOx/G catalyst could be explained by the introduction of Ag nanoparticles which may promote the adsorption of oxygen due to induced defects by forming Ag−O−Mn bonds [59]. On the basis of the onset potential values, the intrinsic ORR activity of the Ag-MnOx/G composite appears to be higher than that of the Ag catalyst.
Degradation Test of Ag-MnOx/G Catalyst
Long-term stability of the prepared catalyst is very important for its application in fuel cells. A repetitive potential cycling was applied to investigate the stability of the Ag-MnOx/G composite toward the ORR. Figure 5 shows the ORR polarization curves before and after 1000 potential scans. Proximity of these RDE voltammetry curves indicates a high stability of the catalyst studied. The half-wave potential (E 1/2) value decreased from 0.72 to 0.68 V vs RHE after the long-term cycling under harsh electrochemical conditions. Ag-MnOx/G composite revealed itself as a stable electrocatalyst for ORR. Inset of Fig. 5 represents a pattern of CV curves during 1000 potential cycles. After each 100 cycles, the area under the CV peak changes fractionally but insignificantly. As indicated by the CVs, the Ag-MnOx/G catalyst showed no loss in silver oxide reduction current and maintained significant durability during repeat potential cycling. High durability of Ag-MnOx/G catalyst was explained by ability to minimize the formation of hydrogen peroxide. Low HO2 ─ production may be due to unique morphological and electronic structures of this composite material. On the one hand, the electron transfer from carbon to MnOx is evident, the resulting positively charged surfaces on the adjacent C atoms would establish favorable sites for the side-on O2 adsorption and facilitate the direct reduction of oxygen to OH─ via a four-electron process [66]. On the other hand, highly dispersed Ag and MnOx nanoparticles with close proximity to each other (approximately <2–3 nm) can provide an ensemble effect acting as a bifunctional catalyst, where these different particles complement each other by catalyzing different oxygen reduction reaction steps (4- and 2-electron reductions), according to previous results for metal–metal oxide composites for ORR [67]. In this way, the HO2 ─ intermediate generated on the Mn3O4 surfaces can easily diffuse to the neighboring Ag surfaces or the Ag–MnOx interface, where it can undergo prompt disproportionation into OH─ and O2 for further oxygen reduction with this hybrid catalyst [68]. Among the formation of H2O2, another reason of activity lost for Ag-MnOx/C catalysts can be related to the formation of manganese carbonates, and their sizes significantly increase to a few nanometer. In addition, their dispersion over the carbon surface is no longer homogeneous. Moreover, the carbon-supported Ag-MnOx nanoparticles are attacked by the alkaline solution. Manganite (MnIII) is soluble in alkaline solution, whereas carbon and silver should be significantly more stable; however, some corrosion of even graphitized carbon might occur in alkaline electrolyte [69]. Dissolution of MnIII (which confers good ORR activity) implicates the decrease of overall electrocatalyst activity [70].

Comparison of RDE polarization curves of oxygen reduction for a Ag-MnOx/G-modified GC electrode before and after 1000 scans in O2-saturated 0.1 M KOH, ν = 10 mV/s, and rotation speed: 1900 rpm. Inset shows long-term CVs of Ag-MnOx/G-modified GC electrode in Ar-saturated 0.1 M KOH. ν = 50 mV/s
Conclusion
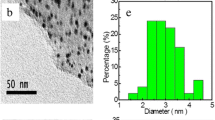
Silver nanoparticles supported on MnOx/graphene composites were prepared using a simple electrodeposition technique. According to physical characterization, Ag nanoparticles with diameter of 4 nm were uniformly deposited over 9–10-nm thick MnOx layer on graphene. The synthesized Ag-MnOx/G nanocomposites demonstrated a higher ORR activity than bulk Ag electrode. The reduction of oxygen on the prepared composite catalyst proceeds via a four-electron pathway in alkaline media, avoiding the formation of hydrogen peroxide. The RDE results show that the oxygen reduction onset potential of the Ag-MnOx/G catalyst shifts positively by 100 and 120 mV compared with that of Ag/graphene and MnOx/graphene composites, respectively. The electrocatalytic activity of the Ag-MnOx/graphene composite improved compared with that of bulk Ag. The RDE results indicate that the Ag-MnOx/graphene composite is a promising cathode catalyst for alkaline membrane fuel cells. Electrodeposited silver nanoparticles significantly improve the electrocatalytic activity of MnOx/graphene nanocomposite by enhancing the conductivity and increasing abundant active sites for the ORR process. The Ag-MnOx/G catalyst showed also a remarkable stability in alkaline media. All these findings demonstrate that the Ag-MnOx/graphene composite would be a promising candidate for non-Pt cathode catalysts in alkaline fuel cells.
References
B. Wang, J. Power Sources 152, 1 (2005)
G. Wu, P. Zelenay, Acc. Chem. Res. 46, 1878 (2013)
P. Trogadas, T.F. Fuller, P. Strasser, Carbon 75, 5 (2014)
G. Lota, K. Fic, E. Frackowiak, Energy Environ. Sci. 4, 1592 (2011)
D.S. Su, R. Schlögl, Chem. Sus. Chem. 3, 136 (2010)
N. Alexeyeva, A. Sarapuu, K. Tammeveski, F.J. Vidal-Iglesias, J. Solla-Gullón, J.M. Feliu, Electrochim. Acta 56, 6702 (2011)
J.-J. Lv, S.-S. Li, A.-J. Wang, L.-P. Mei, J.-R. Chen, J.-J. Feng, Electrochim. Acta 136, 521 (2014)
S.O. Kim, D.H. Lee, W.J. Lee, W.J. Lee, Y.H. Kim, Phys. Rev. Lett. 106, 175502 (2011)
I. Kruusenberg, J. Mondal, L. Matisen, V. Sammelselg, K. Tammeveski, Electrochem. Commun. 33, 18 (2013)
M. Vikkisk, I. Kruusenberg, U. Joost, E. Shulga, I. Kink, K. Tammeveski, Appl. Catal. B Environ. 147, 369 (2014)
S. Ratso, I. Kruusenberg, M. Vikkisk, U. Joost, E. Shulga, I. Kink, T. Kallio, K. Tammeveski, Carbon 73, 369 (2014)
I. Kruusenberg, S. Ratso, M. Vikkisk, P. Kanninen, T. Kallio, A.M. Kannan, K. Tammeveski, J. Power Sources 281, 94 (2015)
N. Alexeyeva, E. Shulga, V. Kisand, I. Kink, K. Tammeveski, J. Electroanal. Chem. 648, 169 (2010)
S. Maldonado, K.J. Stevenson, J. Phys. Chem. B109, 4707 (2005)
K.P. Gong, F. Du, Z.H. Xia, M. Durstock, L.M. Dai, Science 323, 760 (2009)
S. Shanmugam, T. Osaka, Chem. Commun. 47, 4463 (2011)
M. Vikkisk, I. Kruusenberg, U. Joost, E. Shulga, K. Tammeveski, Electrochim. Acta 87, 709 (2013)
M. Piana, S. Catanorchi, H.A. Gasteiger, ECS Trans. 16, 2045 (2008)
F. Cheng, J. Shen, B. Peng, Y. Pan, Z. Tao, J. Chen, Nature Chem. 3, 79 (2011)
N. Ominde, N. Bartlett, X.-Q. Yang, D. Qu, J. Power Sources 185, 747 (2008)
C.-C. Yang, S.-T. Hsu, W.-C. Chien, M.C. Shih, S.-J. Chiu, K.-T. Lee, C.L. Wang, Int. J. Hydrogen Energy 31, 2076 (2006)
M.L. Calegaro, F.H.B. Lima, E.A. Ticianelli, J. Power Sources 158, 735 (2006)
I. Roche, K. Scott, J. Electroanal. Chem. 638, 280 (2010)
J. Wu, D. Zhang, Y. Wang, Y. Wan, Electrochim. Acta 75, 305 (2012)
J. Zhu, J. He, ACS Appl. Mater. Interfaces 4, 1770 (2012)
R. Zou, Z. Zhang, L. Yu, Q. Tan, Z. Chen, J. Hu, Chem. Eur. J. 11, 13912 (2011)
E.M. Benbow, S.P. Kelly, L. Zhao, J.W. Reutenauer, S.L. Suib, J. Phys. Chem. C 115, 22009 (2011)
F. Cheng, Y. Su, J. Liang, Z. Tao, J. Chen, Chem. Mater. 22, 898 (2010)
Y. Ma, R. Wang, H. Wang, J. Key, S. Ji, J. Power Sources 280, 526 (2015)
Y.L. Cao, H.X. Yang, X.P. Ai, L.F. Xiao, J. Electroanal. Chem. 557, 127 (2003)
L. Mao, D. Zhang, T. Sotomura, K. Nakatsu, N. Koshiba, T. Ohsaka, Electrochim. Acta 48, 1015 (2003)
I. Roche, E. Chaînet, M. Chatenet, J. Vondrák, J. Phys. Chem. C 111, 1434 (2007)
J.S. Spendelow, A. Wieckowski, Phys. Chem. Chem. Phys. 9, 2654 (2007)
L. Tammeveski, H. Erikson, A. Sarapuu, J. Kozlova, P. Ritslaid, V. Sammelselg, K. Tammeveski, Electrochem. Commun. 20, 15 (2012)
J. Guo, A. Hsu, D. Chu, R. Chen, J. Phys. Chem. C 114, 4324 (2010)
J.-J. Han, N. Li, T.-Y. Zhang, J. Power Sources 193, 885 (2009)
L. Demarconnay, C. Coutanceau, J.M. Léger, Electrochim. Acta 49, 4513 (2004)
H. Meng, P.K. Shen, Electrochem. Commun. 8, 588 (2006)
N. Wagner, M. Schulze, E. Gulzow, J. Power Sources 127, 264 (2004)
B.B. Blizanac, P.N. Ross, N.M. Markovic, Electrochim. Acta 52, 2264 (2007)
B.B. Blizanac, P.N. Ross, N.M. Markovic, J. Phys. Chem. B 110, 4735 (2006)
Q.-m. Wu, J.-m. Ruan, Z.-c. Zhou, S.-B. Sang, Trans. Nonferrous Met. Soc. China 25, 510 (2015)
Y.G. Sun, Y.N. Xia, Science 298, 2176 (2002)
L.Z. Yuan, L.H. Jiang, J. Liu, Z.X. Xia, S.L. Wang, G.Q. Sun, Electrochim. Acta 135, 168 (2014)
M. Chatenet, L.G. Bultel, M. Aurousseau, R. Durand, F. Andolfatto, J. Appl. Electrochem. 1131, 32 (2002)
A. Fazil, R. Chetty, Electroanalysis 26, 2380 (2014)
C. Coutanceau, L. Demarconnay, C. Lamy, J.M. Leger, J. Power Sources 156, 14 (2006)
P. Singh, D.A. Buttry, J. Phys. Chem. C 116, 10656 (2012)
W. Gac, Appl. Catal. B 75, 107 (2007)
F.-P. Hu, X.-G. Zhang, F. Xiao, J.-L. Zhang, Carbon 43, 2931 (2005)
M.A. Kostowskyj, D.W. Kirk, S.J. Thorpe, Int. J. Hydrogen Energy 35, 5666 (2010)
Q. Tang, L. Jiang, J. Qi, Q. Jiang, S. Wang, G. Sun, Appl. Catal. B104, 337 (2011)
G.-Q. Zhang, X.-G. Zhang, Y.-G. Wang, Carbon 42, 3097 (2004)
I. Roche, K. Scott, J. Appl. Electrochem. 39, 197 (2009)
P. Bezdicka, T. Grygar, B. Klapste, J. Vondrak, Electrochim. Acta 45, 913 (1999)
B. Klapste, J. Vondrak, J. Velicka, Electrochim. Acta 47, 2365 (2002)
J. Vondrak, B. Klapste, J. Velicka, M. Sedlarikova, J. Reiter, I. Roche, E. Chainet, J.F. Fauvarque, M. Chatenet, J. New Mater. Electrochem. Sys. 8, 209 (2005)
J. Chen, X. Tang, J. Liu, E. Zhan, J. Li, X. Huang, W. Shen, Chem. Mater. 19, 4292 (2007)
H. Huang, Y. Meng, A. Labonte, A. Dobley, S.L. Sui, J. Phys. Chem. C 117, 25352 (2013)
L. Sun, Q. Cao, B. Hu, J. Li, J. Hao, G. Jing, X. Tang, Appl. Catal A 393, 323 (2011)
F.H.B. Lima, M.L. Calegaro, E.A. Ticianelli, Electrochim. Acta 52, 3732 (2007)
S. Liu, X. Qin, RSC Adv. 5, 15627 (2015)
A.J. Bard, L.R. Faulkner, Electrochemical Methods, 2nd edn. (Wiley, New York, 2001)
R.E. Davis, G.L. Horvath, C.W. Tobias, Electrochim. Acta 12, 287 (1967)
D.R. Lide (ed.), CRC Handbook of Chemistry and Physics, 82nd edn. (CRC Press, Boca Raton, 2001)
Z. Yang, X. Zhou, H. Nie, Z. Yao, ACS Appl. Mater. Interfaces 3, 2601 (2011)
D.A. Slanac, A. Lie, J.A. Paulson, K.J. Stevenson, K.P. Johnston, J. Phys. Chem. C 116, 11032 (2012)
J. Liu, J. Liu, W. Song, F. Wang, Y. Song, J. Mater. Chem. A 2, 17477 (2014)
P.N. Ross, M. Sattler, J. Electrochem. Soc. 135, 1464 (1988)
I. Roche, E. Chaînet, M. Chatenet, J. Vondrák, J. Appl. Electrochem. 38, 1195 (2008)
Acknowledgments
This research was financially supported by institutional research funding (IUT20-16 and IUT2-24) of the Estonian Ministry of Education and Research. We gratefully acknowledge the financial support provided by the Estonian Research Council (Grant No. 9323).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Shypunov, I., Kongi, N., Kozlova, J. et al. Enhanced Oxygen Reduction Reaction Activity with Electrodeposited Ag on Manganese Oxide–Graphene Supported Electrocatalyst. Electrocatalysis 6, 465–471 (2015). https://doi.org/10.1007/s12678-015-0266-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12678-015-0266-x