
Abstract
Groundwater is the most important source of water supply in Iran and understanding the geochemical evolution of groundwater is important for sustainable development of the water resources in Tabas area. A total of 29 samples of groundwater in Tabas area have been analyzed for ions and major elements. Groundwater of the study area is characterized by the dominance of Na–Cl water type. Groundwater was generally acidic to high alkaline with pH ranging from 5.42 to 10.75. The TDS as a function of mineralization characteristics of the groundwater ranged from 479 to 10,957 mg/l, with a mean value of 2,759 mg/l. The Ca2+, Mg2+, SO4 2− and HCO3 − were mainly derived from the dissolution of calcite, dolomite and gypsum. The Cu, Pb and Zn ions are not mobile in recent pH–Eh, but these conditions controlled dissolved Se, V and Mo in groundwater. The As is released in groundwater as a result of the weathering of sulfide minerals like arsenopyrite.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Groundwater is often the only reliable source of fresh water and it is the only renewable water resource in arid regions and in such area, groundwater is commonly the only water resource (Al-ahmadi and El-Fiky 2009). Groundwater occurs almost everywhere beneath the earth surface not in a single widespread aquifer but in thousands of local aquifer systems and compartments that have similar characters. Knowledge of the occurrence, replenishment and recovery of groundwater has special significance in arid and semi-arid regions due to discrepancy in monsoonal rainfall, insufficient surface waters and over drafting of groundwater resources (Vasanthavigar et al. 2010). Water shortages have become an increasingly serious problem in Iran, especially in the arid and semi-arid regions of eastern, southern and central Iran and in these areas groundwater is the most important source for domestic, industrial and agricultural purpose. Variation in groundwater chemistry is mainly a function of the interaction between the groundwater and the mineral composition of the aquifer materials through which it moves and is controlled by various factors, like evaporation, dissolution–precipitation, weathering of silicates, oxidation–reduction, sorption and exchange reactions, transformation of organic matter and mixing processes (Carrilo-Rivera et al. 2007; Sharif et al. 2008). Hydrogeological and geochemical studies are the basis for a scientific groundwater resource management. Despite importance of groundwater in Iran, little is known about the natural phenomena that govern the chemical composition of groundwater or anthropogenic factors that presently affect them (Jalali 2006). Increased knowledge of geochemical evolution of groundwater in these arid regions could lead to improved understanding of hydrochemical systems in such areas, leading to sustainable development of water resources and effective management of groundwater resource. In this article, we reported the results of hydrogeochemical study at Tabas area and define the principal hydrogeochemical processes controlling groundwater quality, based on the major ion and trace element chemistry.
Study area
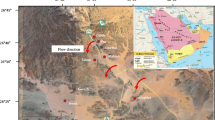
The study area is located around Tabas city, central of Iran (Fig. 1). The Tabas basin is an intermountain depression with an elevation of about 660 m above sea level (a.s.l.). The areas weather is dry, hot and desert. The mean air monthly temperature is highest during August (50 °C) and lowest during January (−5 °C) (Karimi Bavandpur and Hajihosaini 2002). Most months of the year is dry and little rain. Normally, little rain falls in winter and early spring. The vegetation is sparse and scattered thorn bushes. The Tabas area is a part of Central Iranian microcontinent that is a segment of the Iran Plate, which is part of the Cimmerian continent collage (Takin 1972). According to Sengör (1990) this microcontinent, which is a part of a magmatic island arc complex, separated from Gondwana at the beginning of the Triassic period to collide with Eurasia, more specifically with the Turan plate, in the Late Triassic. Sedimentation on the blocks during the Mesozoic was partly shallow marine, partly terrestrial in nature. The Tabas subsidence basin has emerged by drifting of mountains in Eastern and Western of basin in a compression phase from middle Tertiary to former Quaternary as a major Graben and filled by Neocene strata’s and quaternary alluviums (Karimi Bavandpur and Hajihosaini 2002). The geology of study area comprises a thick sequence of sediments of Mesozoic (Triassic) to Quaternary age. The surface geology map shows that 60 % of the land is covered by Quaternary deposits (Fig. 1) and basement rocks in the study area mainly consist of sandstone, conglomerate, limestone and gypsum. In the northeast and eastern of the basin, metamorphic rocks include schist (Precambrian age) being dominant (Aghanabati 1978) and in the southern part, there are shale rocks.

Geology map and groundwater flow direction of the study area (Modified after Aghanabati 1978)
Materials and methods
A total of 29 groundwater samples were collected from existing wells in the study areas in March 2009. The locations of these samples are shown in Fig. 1. Groundwater samples were collected following the standard procedures which included (Halim et al. 2009; Smedley et al. 2002): (1) filtered using 0.45 μm filters and acidified with supra pure 0.7 N HNO3 to pH ∼2 for the analysis of heavy and trace elements; (2) filtered but not acidified for the analysis of major ions and (3) not filtered and not acidified for the analysis of alkalinity. Conductivity, pH, Eh and temperature were determined in the field using portable pH meter. The cations and all trace elements were measured with inductively coupled plasma and mass spectrometry (ICP–MS). The anions (Cl, F, Br, SO4 and NO3) were analyzed using ion chromatography. Total dissolved solids (TDS) were measured by sample evaporation techniques and HCO3 was analyzed by potentiometric determination (Table 1).
Results and discussion
Physicochemical data and major ionic concentrations of 29 analyzed groundwater samples are summarized in Table 1. Groundwater was generally acidic to high alkaline with pH ranging from 5.42 to 10.75. The TDS as a function of mineralization characteristics of the groundwater ranged from 479 to 10,957 mg/l, with a mean value of 2.759 mg/l. Values of the redox potential are in the range −39.4 to 645.1 mV. Martino waters show slightly reducing conditions and demonstrate the presence of a oxidizing to reducing aquifers in the study areas. Among the cations, the concentrations of Ca2+, Mg2+, Na+ and K+ ions ranged from 33.33 to 369.09, 13.41 to 210.98, 72.97 to 3,514.49 and 1.98 to 104.54 mg/l with a mean of 111.15, 53.17, 805 and 8.66 mg/l, respectively. Their concentrations represent on average 11.36, 5.44, 82.31 and 0.89 % of all the cations, respectively. The order of abundance is Na+ > Ca2+ > Mg2+ > K+. Among the anions, the concentrations of HCO3 −, Cl−, SO4 2−, NO3 − ions lie in between 96.81 and 833.22, 70.71 and 3,818.1, 95.71 and 2,802.8, 0 and 59.96 mg/l with a mean of 217.2, 857.42, 679.78 and 14.46 mg/l, respectively. The order of their abundance is Cl− > SO4 2− > HCO3 − > NO3 −, contributing on average, respectively, 48.47, 38.43, 12.28 and 0.82 % of the total anions. Carbonate is not abundant; it represents on average 0.005 % of all the anions. Under arid/semi-arid conditions, strong evaporation would have dramatically affected the groundwater chemistry; therefore, the groundwater from the discharge area contained more Na+ and Cl− (Sikdar et al. 2001; Guo and Wang 2005). The groundwater samples were plotted onto Piper’s diagrams that show the relative concentrations of the different ions from individual water samples (Fig. 2). Two groundwater groups have been identified on the basis of major ion concentrations. The Na–Cl water type is dominated in the most part of the studied area except for two samples (number 4 and 9) that placed in Ca–Mg–SO4–Cl type.

Piper diagram for the groundwater samples in the studied area
To understand the spatial control of major ions concentration, the relationships between TDS (as a useful indicator of anthropogenic contamination) and major ions are illustrated in Fig. 3. The Cl− ion tends to have a stable concentration because it undergoes few chemical and biological reactions in a natural environment, and has, therefore, been widely used as a conservative reference element to study water–rock interactions. Chloride mainly originates from the dissolution of halite in evaporate deposits and the precipitation of dry fallout from the atmosphere (He et al. 2012). The Cl− and Na+ concentrations increased with increasing TDS in all of the groundwater samples (Fig. 3). In arid regions, the build-up of dissolved species through evaporation is a major control on groundwater salinity (Richer and Kreitler 1993; Drever 1997; He et al. 2012), especially for the concentration of Cl− and Na+, which is highly soluble in groundwater. In most groundwater systems, Cl−–Na+ and TDS can be used to represent the intensity of evaporation. This suggests that the dissolution of halite is a considerable control on the Na+ and Cl− concentrations. The TDS and SO4 2− concentrations were strongly and significantly correlated, suggesting that weathering of gypsum (CaSO4·2H2O) occurred in area. Potassium shows weak correlation with TDS and Cl−, indicating that weathering of sylvite (KCl) partly controls the K+ chemistry, other possible sources of K+ in groundwater such as K-feldspars and K-bearing minerals are rare in the studied area (Figs. 1, 3). Study of the bedrock geology of the basin (Fig. 1) suggested that carbonate dissolution was happening mainly in the study area, which increased concentrations of Ca2+ and HCO3 − as:

Relationships between ion concentrations for Na+, K+, Cl−, SO4 2−, Mg2+, Ca2+, HCO3 − and pH with TDS
The dissolution of both gypsum and dolomite will promote calcite supersaturation:
The study area is a coal sedimentary basin in which different coal layers of Triassic–Jurassic age seen (Fig. 1). These layers are in different parts locally or industrial mining. In addition to coal-mining activities happened in this area and in many veins of coal, pyrite has been reported (Zadehkabir 1985). Sulfide minerals, such as pyrite, would have been oxidized when exposed to air due to coal mining in study area, which produced SO4 2−and H+ [Eq. (3)] (Guo and Wang 2005).
The reaction products could be introduced into the groundwater and increase the concentration of SO4 2− and decrease pH of the groundwater in this zone. Low pH caused the following reaction to proceed to the right (Guo and Wang 2005).
This process decreased the concentration of CO3 2− and caused an under-saturated state with respect to calcite and dolomite in the groundwater and promoted dissolution of calcite and dolomite in the aquifer(Guo and Wang 2005), which in turn increased the concentrations of Ca2+ and Mg2+(Fig. 4).

Plot of Mg2+ and Ca2+ versus SO4 2− in groundwater for study area
Concentrations of Si are in the range 5.05–13.12 mg/l (Table 2). Many of the silicate minerals in the aquifer are fine-grained (e.g., clays) and easily weathered. Reaction of silicate minerals is also a key process responsible for the generation of high-pH groundwater. Reaction of sodic feldspar, for example, consumes protons and results in a pH rise. Reaction of albite (NaAlSi3O8) to form kaolinite (Al2Si2O5(OH)4) can be described as (Smedley et al. 2002):
The reaction also accounts for the origin of high Na concentrations in the groundwater (Smedley et al. 2002) (Fig. 5).

Plot of pH and Na+ versus Si in groundwater for study area
Trace metals
Concentration of metals in groundwater is presented in Table 2. Arsenic concentrations in the samples ranged from 1.3 to 31.3 μg/l and six samples were above the WHO drinking water (WHO 2004) guideline for As (of 10 μg/l). In many example, arsenic that form oxyanions is released from Fe and Mn oxides by desorption (Acharyya et al. 2000; Smedley et al. 2002; Mukherjee et al. 2008). The concentrations of Fe and Mn in the studied groundwater samples show a distinct relationship with the concentration of As (Table 2; Fig. 6). The lack of correlation between dissolved As and both Fe and Mn in the groundwater may result from the precipitation of secondary Fe and Mn-mineral phases like siderite (FeCO3), vivianite [Fe3 (PO4)28H2O], pyrite (FeS2) and rhodochrosite (MnCO3) (McArthur et al. 2001; Aziz Hasan et al. 2009). A good positive correlation between As and SO4 2− suggests that As is released in groundwater as a result of the weathering from sulfide minerals like arsenopyrite (FeAsS) (Harvey et al. 2005). A distinct correlation between As and HCO3 − in the groundwater samples (Fig. 6) further suggests mobilization of As in the water in anoxic environment (Halim et al. 2009). A good positive correlation between As and pH how that arsenic is more soluble in an alkaline environment (Fig. 6).

Plot of pH, SO4 2−, HCO3 − and Mn versus As in groundwater for study area
Copper, Zn and Pb concentrations are below the WHO guideline value and not correlated with pH and Eh. Copper concentrations are in the range 0.4–6.9 μg/l, whereas Pb concentrations are usually below the detection limit (0.1 μg/l) and concentrations of Zn show variation between <0.5 and 49.1 μg/l. Elements such as Ag, Cd, Bi and Hg are mostly below detection limits in the waters. Selenium concentrations were found to be between 1.1 and 21.3 μg/l with two samples (sample 28 and 13) measured exceeding the WHO guideline value of 10 μg/l. Selenium, V and Mo transformation and transitioning in natural aquatic systems are influenced by site specific hydrogeochemical and biological processes including the source and removal pathways, biological productivity in the water and sediment compartments and alkalinity and oxidization potential (Somay et al. 2008). Selenium occurs in oxidizing solutions as selenite (SeO −23 ) or selenate (SeO −24 ) ionic species in the groundwater (Hem 1985). Vanadium occurs mainly as trace constituent in pyroxenes, amphiboles; micas and apatite even if can originate from some own minerals. It has three main oxidation states: +3, +4 and +5. Vanadium is a highly mobile element, whose solubility is strongly controlled by its oxidation state. It displays the highest solubility in oxic environments, where vanadate ions (e.g., H2VO4 −) typically dominate the water chemistry (Dinelli et al. 2012). Dissolved Se, V and Mo speciation are a function of salinity, pH and Eh in Tabas area (Fig. 7). Similar associations have been found in saturated soils from the San Joaquin Valley, California (Fujii et al. 1988). Barium concentrations are relatively low, with an observed range 10.48–78.73 μg/l. Concentrations are limited by barite solubility. Bromide concentrations are also high in some groundwater (Table 2), varying between 21 and 4,010 μg/l. Concentrations are highest in the more saline samples and Br correlates well with Cl. The increases are therefore likely to be caused by evaporation (Fig. 8).

Plot of Se, V and Mo versus Eh, E.C, pH in groundwater for study area

Plot of Br versus Cl in groundwater for study area
Summary and conclusions
In the present study, an attempt was made to understand the geochemical evolution of groundwater in the Tabas basin using hydrogeochemical data. Based on the analysis of geochemical data conceptual hydrochemical models were constructed. The work shows that the hydrogeochemistry of the groundwater in study area is mainly controlled by water–rock interaction. Groundwater is mostly Na–Cl type with Cl− as the dominant anion, Na+ as the dominant cation and only two sample of other type of water observed. The high salinity values are a problem in arid regions and are generated by evaporation and this may also be partially responsible for accumulation of some trace elements. The dissolution of halite is a considerable control on the Na+ and Cl− concentrations and weathering of gypsum (CaSO4·2H2O) occurred in area as SO4 2− concentrations. The Ca2+, Mg2+, SO4 2− and HCO3 − were mainly derived from the dissolution of calcite, dolomite and gypsum. Carbonate rocks are present at all of the area, but they may contribute to acid buffering produced by the oxidation of sulphide minerals, especially pyrite. The weathering of silicate minerals also appears able to reduce the acidity. The lack of correlation between dissolved As and both Fe and Mn in the groundwater and good positive correlation between As with pH, SO4 2− and HCO3 − suggests that As is released in groundwater as result of the weathering of from sulfide minerals and arsenic is more soluble in an alkaline and anoxic environment. The recent pH–Eh conditions controlled dissolved Se, V and Mo in groundwater and Cu, Pb and Zn ions are not mobile.
References
Acharyya SK, Lahiri S, Raymahashay BC, Bhowmik A (2000) Arsenic toxicity of groundwater of the Bengal Basin in India and Bangladesh: the role of Quaternary stratigraphy and Holocene sea-level fluctuation. Environ Geol 39:1127–1137
Aghanabati A (1978) Geological map of Iran 1:250,000 series-Tabas. Geological Survey of Iran, Iran
Al-ahmadi ME, El-Fiky AA (2009) Hydrogeochemical evaluation of shallow alluvial aquifer of Wadi Marwani, western Saudi Arabia. J King Saud Univ (Sci) 21:179–190
Aziz Hasan M, Bhattacharya P, Sracek O, Matin Ahmaed K, Bromssen M, Jacks V (2009) Geological controls on groundwater chemistry and arsenic mobilization: hydrogeochemical study along an E–W transect in the Meghna basin, Bangladesh. J Hydrol 378:105–118
Carrilo-Rivera JJ, Varsanyi I, Kovacs LO, Cardona A (2007) Tracing groundwater flow systems with hydrogeochemistry in contrasting geological environments. Water Air Soil Pollut 184:77–103
Dinelli E, Lima A, Albanese S, Birke M, Cicchella D, Giccio L, Valera P, De Vivo B (2012) Major and trace elements in tap water from Italy. J Geochem Explor 112:54–75
Drever JI (1997) The Geochemistry of Natural Waters: Surface and Groundwater Environments, 3rd edn. Prentice Hall, New Jersey
Fujii R, Deverel SJ, Hatfield DB (1988) Distribution of selenium in soils of agricultural fields, western San Joaquin Valley, California. Soil Sci Soc Am J 52:1274–1283
Guo H, Wang Y (2005) Geochemical characteristics of shallow groundwater in Datong basin, northwestern China. J Geochem Explor 87:109–120
Halim MA, Majumder RK, Nessa SA, Hiroshiro Y, Uddin MJ, Shimada J, Jinno K (2009) Hydrogeochemistry and arsenic contamination of groundwater in the Ganges Delta Plain, Bangladesh. J Hazard Mater 164:1335–1345
Harvey CH, Swartz C, Badruzzaman ABM, Keon-Blute N, Yu W, Ali MA, Jay J, Beckie R, Niedan V, Brabander D, Oates P, Ashfaque KN, Islam S, Hemond HF, Ahmed MF (2005) Groundwater arsenic contamination on the Ganges delta: biogeochemistry, hydrology, human perturbations, and human suffering on a large scale. C R Geosci 337:285–296
He J, Ma Jinzhu, Zhang P, Tian L, Zhu G, Edmunds WM, Zhang Q (2012) Groundwater recharge environments and hydrogeochemical evolution in the Jiuquan Basin, Northwest China. Appl Geochem 27:866–878
Hem JD (1985). Study and interpretation of the chemical characteristics of natural water, 3rd edn. USGS Water-supply paper 2254, USA
Jalali M (2006) Chemical characteristics of groundwater in parts of mountainous region, Alvand, Hamadan, Iran. Environ Geol 51:433–446
Karimi Bavandpur A, Hajihosaini A (2002). Geological map of Iran 1:100,000 series-Tabas. Geological Survey of Iran, Iran
McArthur JM, Ravenscroft P, Safiullah S, Thirlwall MF (2001) Arsenic in groundwater: testing pollution mechanisms for aquifers in Bangladesh. Water Resour Res 37:109–117
Mukherjee A, Bromssen MV, Rcanlon BR, Bhattacharya P, Fryar AE, AzizHasan M, Ahmed KM, Chatterjee D, Jacks G, Sracek O (2008) Hydrogeochemical comparison and effects of overlapping redox zones on groundwater arsenic near the Western (Bhagirathi sub-basin, India) and Eastern (Meghna sub-basin, Bangladesh) margins of the Bengal Basin. J Contam Hydrol 99:31–48
Richer BC, Kreitler CW (1993) Geochemical Techniques for Identifying Sources of Groundwater Salinization. CRC Press, Boca Raton
Sengör AMC (1990) A new model for the late Palaeozoic-Mesozoic tectonic evolution of Iran and implications for Oman. In: Robertson AHF, Searle MP, Ries AC (eds) The Geology and Tectonics of the Oman Region, 37. Publication, Geological Society London Special, pp 797–831
Sharif MU, Davis RK, Steele KF, Kim B, Kresse TM, Fazio JA (2008) Inverse geochemical modeling of groundwater evolution with emphasis on arsenic in the Mississippi River Valley alluvial aquifer, Arkansas (USA). J Hydrol 350:41–55
Sikdar P, Sarkar S, Palchoudhwy S (2001) Geochemical evolution of groundwaters in the Quaternary aquifer of Calcutta and Howrah, India. J Asian Earth Sci 19:579–594
Smedley PL, Nicolli HB, Macdonald DMJ, Barros AJ, Tullio JO (2002) Hydrogeochemistry of arsenic and other inorganic constituents in groundwaters from La Pampa, Argentina. Appl Geochem 17:259–284
Somay AM, Gemici U, Filiz S (2008) Hydrogeochemical investigation of Kucuk Menderes River coastal wetland, Selcuk–Izmir, Turkey. Environ Geol 55:149–164
Takin M (1972) Iranian geology and continental drift in the Middle East. Nature 235:147–150
Vasanthavigar, M., Srinivasamoorthy, K., Vijayaragavan, K., Rajiv Ganthi, R., Chidambaram, S., Anandhan, P., Manivannan, R., Vasudevan, S., 2010. Application of water quality index for groundwater quality assessment: Thirumanimuttar sub-basin, Tamilnadu, India. Environ Monit Assess. doi:10.1007/s10661-009-1302-1
WHO (2004) Guidelines for drinking water quality, vol 1 recommendations, 3rd edn. WHO, Geneva
Zadehkabir A (1985) Origin of sulphur contents of coal seams in Parvardeh deposit (Iran). National Steel Corporation (NISCO), Tehran, Iran
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pazand, K., Sarvestani, J.F. Hydrogeochemical investigation in an arid region of Iran (Tabas, Central Iran). Environ Earth Sci 70, 743–752 (2013). https://doi.org/10.1007/s12665-012-2162-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12665-012-2162-5