
Abstract
Elevated concentrations of arsenic in the sediment and pore water in the Sundarban wetlands pose an environmental risk. Adsorption and desorption are hypothesized to be the major processes controlling arsenic retention in surface sediment under oxic/suboxic condition. This study aims to investigate sorption kinetics of As(III & V) and its feedback to arsenic mobilization in the mangrove sediment. It ranges from sand to silty clay loam and shows the adsorption of As(III & V) following the Langmuir relation. Estimates of the maximum adsorption capacity are 59.11 ± 13.26 μg g−1 for As(III) and 58.45 ± 8.75 μg g−1 at 30°C for As(V) in the pH range 4 to 8 and salinity 15–30 psu. Extent of adsorption decreases with increasing pH from 4 to 8 and desorption is the rate-limiting step in the reaction of arsenic with sediment. Arsenic in the sediment could be from a Himalayan supply and co-deposited organic matter drives its release from the sediment. Arsenic concentration in the sediment is well below its maximum absorption capacity, suggesting the release of sorbed arsenic in pore water by the microbial oxidation of organic matter in the sediment with less feedback of adsorption.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The mobility and fate of arsenic (As) either in the sediment or aquatic environment are controlled by (a) redox condition (b) dissolution–precipitation of mineral with arsenic as a constituent ion and (c) sorption–desorption. Hering and Kneebone (2002) reported that solubilization of only 0.09% of the total arsenic (1.8 mg kg−1) from crustal rock is required for the abundance of 10 μg L−1 of arsenic in water.
Precipitation of authigenic minerals with arsenic as a constituent ion is unlikely, as most arsenic minerals are too soluble to precipitate under oxic condition (Wagemann 1978; Webster 1990; Bowell 1994). However, dissolution and precipitation of authigenic arsenic minerals are kinetically slow process and may be controlled by microbial activity (Das et al. 1996; Zobrist et al. 2000; Acharya et al. 2000). Recent studies, however, have demonstrated that the distribution of sulfate reduction may in fact be the most important factor in determining whether arsenic can accumulate to hazardous levels in groundwater in naturally contaminated reducing aquifers (Kirk et al. 2004; Buschmann and Berg 2009). Authigenic arsenopyrite produced during sulfate reduction is the most important stable iron-sulfide sink for arsenic in these systems (Lowers et al. 2007). Unlike authigenic mineral precipitation, sorption/oxidative or reductive desorption may be an important process in controlling the dissolved arsenic concentrations. Different affinities of As(III) and As(V) can affect the extent of their sorption in the sediment. Varying capacity for As sorption into the sediment may ultimately affect its concentration in the ambient/water. Spatial variability of dissolved As found in shallow aquifers of the Gangetic Delta remains poorly understood (Mertal et al. 2008). These arsenic bearing aquifers are mainly confined to sediments discharged from the rapidly eroding Himalaya, approximately 7,500–10,000 years ago (Acharya et al. 2000; Goodbred and Kuehl 2000). So, arsenic occurring in the sediment of Gangetic Delta is from Himalayan source. Apart from this, suspended sediment could scavenge arsenic from sea water before its deposition in the mangrove system. Banerjee et al. (2011) reported the mass accumulation rates of 0.41–0.66 g cm−2 year−1 determined from 210Pb geochronology of core sample and concluded that variations in trace metal content with depth result largely from their input owing to anthropogenic activities rather than diagenetic process. Our previous study on the spatial and temporal variation of arsenic in the Indian Sundarban mangrove ecosystem (Mandal et al. 2009) showed significant diagenetic remobilization of arsenic with a dispersal rate between 80.7 and 509 ng m−2 d−1. High concentration of arsenic and two benchmarks of arsenic [threshold effect level (TEL) and effects range-low (ERL)] observed in the sediment cores from Sundarban appear to be associated with a potential biological risk (Silva Filho et al. 2010). Highly productive Sundarbans mangrove ecosystem (4.71–6.54 Mg C ha−1 year−1) with 4.85 Mg C ha−1 year−1 of litter production (Ray et al. 2011) could be a source of organic matter which could accelerate the microbial conversion from oxic to suboxic condition in the deeper layer of sediment column. Therefore, availability of Fe(III) oxy hydroxide in the sediment could control sequestration and release of arsenic depending on its diagenetic alteration under changing redox condition (Acharya et al. 2000). Mandal et al. (2009) suggest that arsenic could be immobilized during incorporation into the arsenic-bearing initial phase, and unlikely to be released into pore water until mangrove derived organic matter fuels microbial reduction of adsorbate and adsorbent in the Sundarban mangrove ecosystem. It needs to answer the guiding question: how important are potential back-reaction, e.g. arsenic adsorption? The purpose of this study is to investigate sorption kinetics of As(III) and As(V) and its feedback to arsenic mobilization in the mangrove sediment with naturally occurring source of arsenic from a Himalayan supply.
Materials and methods
Sampling and sample preparation
During 23 March, 2008, mangrove water (low and high tide) and four sediment cores were obtained from four stations (Fig. 1) located in the Indian Sundarban at 21°32′, 22°40′ N and 88°05′, and 89°E by pressing a pre-cleaned acrylic tube (5.5 cm i.d.) in the bed and carefully retrieving it. Average maximum corer penetration of 30 cm was possible due to sediment compaction root network beyond this point. The sediment contained in the acrylic tube was carefully pushed out onto a clean glass plate using an acrylic plunger and sectioned at every 4–5 cm interval. Then the sediment was wet sieved (63 μm) and subsequently air dried/freeze dried and agglomerates were broken in a mortar and pestle. Pore water was separated by centrifugation (30 min, 5,000 rpm). Tidal and pore water samples were filtered (0.45 μm) and stabilized by the addition of H2SO4 (0.1% v/v).

Map showing the station locations
Laboratory procedures
pH was measured using a combined electrode (Systronics, micro pH system 362) and redox potential profiles were measured by gently lowering the electrodes into the sediment (Vischer et al. 1991). Salinity of the samples was determined by Mohr–Knudsen titration. Freeze dried sediment sample was used for the measurement of organic carbon by the Walkey–Black wet combustion method (Shrawat 1982). Particulate matter filtered from the tidal water and sediment were freeze dried and the extracts obtained after treating them with acid mixture (HF/HNO3) were used for the measurement of As. Air dried sediment sample was used for grain size analysis following the sedimentation method (Sims and Heckendorn 1991). Iron in the soil was determined by AAS after acid digestion. Non lithogenous fraction of CaCO3 was determined by the extraction with 1 M hydroxyl amine hydrochloride and 25% acetic acid mixture followed by the analysis of calcium using the method of Culkin and Cox (1966).
Fulvic and humic acids were determined by the KOH (0.1 N) extraction by using Perkin-Elmer LS-50 luminescence spectrophotometer for fluorescence measurement against the standard [excitation and emission at 313 and 425 nm for fulvic acid and 392 and 484 nm for humic acid (Ghatak et al. 2002)].
Adsorption kinetic studies were carried out in a temperature controllable water bath at 20, and 30°C in nitrogen atmosphere using 0.5 g of freeze dried sediment (Lothian) in 100 ml of artificial sea water at salinity 15, 30 psu and pH 4, 6, and 8. The doped artificial sea water was prepared by mixing artificial sea water with 1,000 μg ml−1 stock solution of arsenic to produce a solution 3,000 μg L−1. Aliquots of the sample (~5 ml) were collected at around 2, 5, 15, 30, 60, 120, 240, 480, 600, 1,440, 1,680 and 1,920 min using 10 ml syringe and filtered through 0.2 μm syringe filter. As concentration in the filtered samples were analyzed using the Varian Hydride system-vapour generator (serial no. ELO 405-314) coupled to the spectra AA55B true double beam atomic absorption spectrometer (Yamamoto et al. 1985; Loring and Rantala 1992). Measurement of As(III) was performed at pH 4 using 5% potassium bi-phthalate (Barman et al. 1977). The analytical methods for arsenic were verified before analyses of sample against the standard sample procured from Merck KGaA, Germany. Relative accuracy and the coefficient of variation were 96.2 and 9.2%, respectively, for arsenic. Samples for the determination of adsorption isotherms were prepared by adding 0.5g of sediment (Lothian) to 100 ml of artificial sea water at a salinity of 15 psu and pH 4. Initially 0.5–20 mg L−1 of Cu(II), Co(III), Zn(II) and 0.4–3 mg L−1 of As(III & V) and P(V) were taken in seawater and the mixture was kept at 30°C for 24 h in nitrogen atmosphere to attain equilibrium. Concentrations of Co, Cu, and Zn in the filtered solution were determined by AAS and P(V) by spectrophotometric method (Grasshoff et al. 1983). Both adsorption isotherm and adsorption kinetic studies were conducted in the nitrogen atmosphere for As(III).
Application of Langmuir adsorption isotherm
The relation between the amount adsorbed by the adsorbent (sediment) and the concentration of the adsorbate at a specified condition is known as the adsorption isotherm. The adsorption data obtained from the isotherm experiments were fitted to Langmuir adsorption isotherm:
where C s is the adsorbate concentration in sediment (μg g−1) and C w is the adsorbate concentration in water (μg L−1); Γm is the maximum adsorbate concentration (μg g−1). K is the equilibrium constant (L μg−1) for the adsorption, and K d is the partition coefficient (C s/C w in L kg−1). The reaction between cations/anions (X) and a single sorption phase, in which the ionic charge has been omitted, can be expressed as follows:
where K ≡ S−X is the conditional stability constant for the reaction, ≡ S−X denotes the total cations/anions concentration sorped by the sediment, [≡ S] denotes the concentration of binding sites and [X] is the equilibrium concentration of cations/anions in water.
Partition coefficient (K d) can be expressed as:
C x denotes the corresponding concentration of cations/anions in sediment and in water, respectively.
Considering two classes of sorption sites for anions/cations such as iron-oxide FeOx and organic matter (OM) and from Eqs. 3 and 4 the following relation for partition coefficient (K d ) is obtained:
The free energy change of adsorption is calculated from the equation: ΔG = −RT ln K d, where R (8.3145 J/mol K) is the ideal gas constant, and T (K) is the temperature.
Rate constant for adsorption (K 1) and desorption (K 2) were calculated from K (equilibrium constant for the adsorption, \( K = \, K_{ 1} /K_{ 2} \). K was calculated using the following equation
\( 1/C \, = { 1}/C_{0} + \, K_{ 1} t/C_{0} \) , where C 0 denotes initial concentration and C after adsorption of the adsorbate at time t.
Results and discussion
Soil characteristics
A summary of sediment characteristics is presented in Table 1. The sediment is of sand to silty clay loam type and the clay content varied from 1.3 to 21.5%. The sediment was composed of quartzo-feldspathic minerals (quartz, albite, microcline) contributed from the eroded rocks of acidic composition of the drainage basin. Flaky mica and chlorite were also found to occur in the sediment, though inconsistent (Sarkar et al. 2004). Organic matter exhibited lower concentration (3.15–5.14 mg g−1) than in the sediment from south east/west coast of India (Kumar 1996; Jonathan and Ram Mohan 2003). This could be due to the marine sedimentation and mixing processes at the sediment water interface where the rate of delivery, as well as rates of degradation by microbial-mediated processes are expected to be high (Canuel and Martens 1993). A downward decrease of organic carbon (0.76 to 0.42%) and Eh (−32.14 to −220.2 mV) are common features for all the stations (Fig. 2), indicating microbial-mediated oxidation (Canuel and Martens 1993) of organic matter. A sediment accumulation rate of 0.3–0.48 cm year−1 (Banerjee et al. 2011) and elevated concentration of arsenic in the deeper layers relative to the surface (2.11–3.1 mg kg−1) indicating that arsenic supply seems to have declined, since the commissioning of Farakka barrage in 1975 to regulate the fresh water flow through the Hooghly–Bhagirathi channel for navigation.

Depth profile of a As (mg kg−1), b organic C (%), c Eh (mV)
Adsorption isotherm
A typical example of adsorption isotherm (plot of C s, concentration in sediment versus C w, concentration in artificial sea water) for As(V) (solid line) and As(III) (dotted line) at pH 4, salinity 15 psu and temperature 20 and 30°C are given in Fig. 3a. Extent of adsorption was increased with increasing As concentration in sea water and adsorption isotherms belong to Langmuir type (convex upward) suggesting low affinity specific adsorption. Plots of C w/C s versus C w yielded straight line (Fig. 3b) indicating equilibrium form of Langmuir model and Langmuir coefficients of mangrove soil are given in Table 2. The mean values of maximum adsorption concentration, Γm (μg g−1) were found to be 53.56 ± 13.86 at 20°C, 59.11 ± 13.26 at 30°C for As(III) and 54.21 ± 10.41 at 20°C, 58.45 ± 8.75 at 30°C for As(V) in the pH range 4–8 and S 15 to 30 psu. Estimates of Γm for sediment samples (organic matter 0.16%, CaCO3 1.7%, clay 51.3%) collected from Tulare Lake, San Joaquin Valley, California, USA showed a decrease from 92 μg g−1 at pH 7.5 to 70 μg g−1 at pH 8.5 (Gao et al. 2006). Siegel et al. (2008) studied partitioning of As between granular ferric oxide and Socorro ground water and found maximum sorption capacity of 1,500 μg g−1of oxide phase at 15°C. Arthur (1984) showed sorption capacity for the sediment of the type silty sand with little clay could vary widely between 1.0 and 252 μg As g−1. Wauchope and McDowell (1984) reported sorption of 3.97μM arsenate onto the sediments collected from alluvial flood plain of the Mississippi River. Data obtained for adsorption of As(III & V), P(V), Co(III), Cu (II) and Zn(II) onto mangrove sediment at pH 4, temperature 30°C and salinity 15 psu are given in Table 3 for comparison. Γm was found greater for cations i.e. Cu(II), Co(III), and Zn(II) than anions i.e. P(V), As(III) and As(V). The sediment exhibited greater adsorption for P(V) than As(V & III).

a Adsorption isotherm (Cs, concentration in sediment versus C w, concentration in artificial sea water) for As(V) (solid line) and As(III) (dotted line) b Plots of C w/C s versus C w at temperature 20, 30°C and pH 4, salinity 15 psu
Adsorption isotherm for both As(III) and (V) is a typical isotherm (Fig. 2a) often found for chemical adsorption. Adsorption of both As(III) and As(V) at pH 4 decreased with increasing salinity which could be due to positive potential (point of zero salt effect, PZSE >point of net zero charge, PZC) in the plane of adsorption (Bolan et al. 1986). The extent of absorptions onto the sediment increased with increasing concentration of arsenic in the liquid phase till it reached a limiting value corresponding to its maximum capacity. The adsorption maxima Γm was found to increase with temperature rise, while bonding energy, K was greater for As(V) than As(III), suggesting that mangrove sediments conform to Langmuir adsorption with a monolayer adsorption site exhibiting greater affinity to As(V) relative to As(III). However, in both cases it can be regarded as ligand exchange reaction with different degree of incorporation into the surface. Low affinity specific adsorption in the sediment could induce additional negative charge by cations (Cu, Co, Zn) for proton desorption and additional positive charges by anions (P, As) (Mott 1981).
Adsorption kinetics
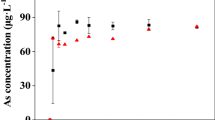
Adsorption kinetics of As(III) and As(V) are given in Fig. 4. Adsorption was found to increase gradually with time till a limiting value was reached. The rate of arsenate adsorption was greater than arsenite and both obeyed a first order kinetics.

Adsorption kinetics for As(V) and As(III) at a pH 4, b pH 6, c pH 8, at salinity 15, 30 psu and temperature 30°C
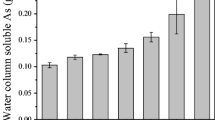
Figure 5 represents the plot of 1/C w versus t for As(V) and As(III) at S 15, pH 4 and temperature 30°C and the curves are resolved into two linear portions representing two distinct types of adsorption sites. Adsorption rate (K 1) is faster in the first linear site I than in the site II for both As(V) and As(III). However, As(V) exhibits faster adsorption rate than As(III) in the site I. (Table 2). The rate constants (K 2) for As(V) and As(III) desorption from sediment are lower than the rate constants (K 1) for adsorption, indicating that desorption is the rate-limiting step. This is in consistent with the hysteresis phenomenon observed for sediment which show the kinetically controlled desorption or irreversible adsorbtion of natural organic matter (Lu and Pignatello 2002), heavy metals such as Cu and Zn (Wu et al. 1999, Pan et al. 1999), and oxyanions such as P (Villapando and Graetz 2001). Goethite (Grossl et al. 1997), similarly, exhibits desorption as the rate-limiting step in the reaction with arsenate and Fuller et al. (1993) suggested that rate limiting desorption of arsenate from ferrihydrite could be limited by diffusional processes. Variation of Γm, maximum adsorbate (As) concentration with respect to pH change is given in Fig. 6. A decrease in adsorption with increase of pH from 4 to 8 was observed for both As(III) and As(V). pH seems to be the most prominent factor in controlling the partitioning of As(III & V) to sediment (Sposito 1984), and Goldberg (2002) suggested that the decrease of arsenate adsorption with increasing pH could be attributed to the occurrence of oxide and clays with less CaCO3 content in the sediment. Hydrogen ion could affect the surface charge of the sediment (adsorbent) and the degree of ionization and the speciation of the adsorbate (Xu et al. 1988). As(III & V) speciation is pH dependent (National Institute of Standards and Technology 2003) and proton bearing surface functional groups on metal oxy hydroxide, clay minerals, and organic material could also undergo pH dependent dissociation. Although the pK1 value of P(V) is 2.12 its Γm was found fivefold greater than As(V). Equilibrium constant for P(V) adsorption was about 300–1,000 times greater than that of As(III) and As(V). The reason is that the arsenate and arsenites formed in the Fe(III) oxide phase (Manceau 1995) are more soluble than that formed by phosphate (Parfitt et al. 1975) and the order of free energy change is As(III) < As(V) < P(V) (Table 3). Authigenic mineral precipitation of arsenate is unlikely in the surface sediment (Langmuir et al. 2006). The values of ΔG calculated at 30°C are negative, indicating that the adsorption is a spontaneous process and bonding forces are strong enough to break the potential barrier. However, ΔG is more negative for As(V) relative to As(III), implying that arsenite is more mobile than arsenate.

A plot of first order kinetic study of As(III) and As(V) on the sediment at pH 4, S 15 psu and temperature 30°C

Variation of Γm, maximum adsorbate (As) concentration (μg g−1) with respect to pH change for a As(V) and b As(III)
Therefore, at high pH values, As(III) and As(V) are adsorbed in a lower extent and at low pH (i.e.4) Γm is greater for Cu(II), Co(III), Zn(II) than P(V), As(V) and As(III), suggesting that the surficial adsorption sites are principally composed of organic matter (humic and fulvic acids) via ligand exchange reaction and adsorption of As(V) and As(III) occurs in both organic matter and hydroxide phase. Lee et al. (1996) made equilibrium batch measurements of the adsorption of Cd(II) on 15 New Jersey soils and observed maximum sorbed concentration ranging from 714 to 6,469 μg g−1 mainly onto the soil organic matter at pH 4, 5 and 6. Lower equilibrium constants (K) for As(III) and As(V) relative to P(V) indicate that slower adsorption and faster desorptions by the underlying metal oxide phase, and reverse is the case for P(V), being controlled by diffusion process (Dzombak and Morel 1990).
Stepwise multiple regression analysis supports two site (Organic matter and Fe-oxide phase) hypothesis for As adsorption in the sediment and important predictors for the variation of As in the sediment are in the decreasing order of importance of the (1) sites: organic carbon, OC% (21.4%), Fe, mg kg−1 (15.5%) (2) ambient physico–chemical parameters: pH (32.5%), T (3.4%), salinity, S psu (0.5%) (As = 10.2 + 4.2 OC + 0.00613 [Fe] − 0.266 S + 0.088T–1.58 pH, R2 = 73.3%, p = 0.16, n = 12). Significant contribution organic carbon for the variation of arsenic concentration in core profiles supports the view (Meharg et al. 2006) that arsenic is codeposited with organic carbon in Bengal delta sediments, and on burial of the sediment degradation of organic matter provide the reducing conditions to dissolve Fe(III) oxides and release arsenic into the pore water. Concentration of As in the mangrove sediment (2.10–3.10 μg g−1) being lower than its concentration corresponding to maximum adsorption capacity indicates release of arsenic to pore water by microbial oxidation of organic matter (Jones et al. 2000) with less feedback of adsorption.
Conclusions
Studies on adsorption–desorption behavior of As(V) and As(III) in the Sundarbans mangrove sediment using Langmuir adsorption isotherm showed that (1) observed concentration of arsenic in the sediment is well below the concentration corresponding to its maximum adsorption capacity which decreases for increasing pH from 4 to 8, and desorption is the rate limiting step in the reaction of As (III) & (V) with the sediment (2) a steady increase in the arsenic concentration with depth in cores could be the result of modified river flow after the commissioning of Farakka barrage in 1975 (3) arsenic and organic matter could be co-deposited and microbial degradation of organic matter drives arsenic release from the sediment to pore water with less feed back of adsorption leading to its migration from sediment to overlying water.
References
Acharya SK, Lahiri S, Kaymahashay BC, Bhowmic A (2000) Arsenic toxicity in ground water in parts of Bengal Basin in India and Bangladesh: role of quaternary stratigraphy and Holocene sea-level fluctuation. Environ Geol 39(10):1127–1137
Arthur DL (1984) Full-scale field evaluation of waste disposal from coal-fired electric generating plants, Final Report to US EPA. Office of Research and Development, Washington, Appendix F (Part 4), EPA contract no. 68-02-3167
Barman RS, Johnson DL, Foreback CC, Ammous JM, Bricker JL (1977) Separation and determination of nanogram amount of inorganic arsenic and metal arsenic compounds. Anal Chem 49(4):621–625
Banerjee K, Senthilkumar B, Purvaja R, Ramesh R (2011) Sedimentation and trace metal distribution in selected locations of Sundarbans mangroves and Hooghly estuary, northeast coast of India. Environ Geochem Health. doi:10.1007/s10653-011-9388-0
Bolan NS, Syers JK, Tillman RW (1986) Ionic strength effects on surface charge and adsorption of phosphate and sulphate by soils. Eur J Soil Sci 37:379–388
Bowell RJ (1994) Sorption of arsenic by iron oxides and oxyhydroxides in soils. Appl Geochem 9:279–286
Buschmann J, Berg M (2009) Impact of sulfate reduction on the scale of arsenic contamination in groundwater of the Mekong, Bengal and Red River Deltas. Appl Geochem 24(7):1278–1286
Canuel EA, Martens CS (1993) Seasonal variability in the sources and alteration of organic matter associated with recently deposited sediments. Org Geochem 20(5):563–577
Culkin F, Cox RA (1966) Sodium potassium, magnesium, calcium and stroncium in sea water. Deep Sea Res 13:785–789
Das D, Samanta G, Mondal BK, Chowdhury RT, Chanda CR, Chowdhury PP, Basu GK, Chakrabarty D (1996) Arsenic in ground water in six districts of West Bengal, India. Environ Geochem Health 18:5–15
Dzombak DA, Morel FMM (1990) Surface complexation modeling: hydrous ferric oxide. Wiley, New York
Fuller CC, Davis JA, Waychunas GA (1993) Surface chemistry of ferrihydrite: part 2. Kinetics of arsenate adsorption and coprecipitation. Geochim Cosmochim Acta 57:2271–2282
Gao S, Goldberg S, Herbel MJ, Chalmers AT, Fujii R, Tanji KK (2006) Sorption processes affecting arsenic solubility in oxidized surface sediments from Tulaure Lake Bed, California. Chem Geol 228:33–43
Ghatak H, Mukhopadhyay SK, Biswas H, Sen S, Jana TK (2002) Quantitative study of CO (III) complexation by synchronous fluorescence spectroscopy with Sundarban mangrove habitat humic substances. Indian J Mar Sci 31(2):136–140
Goldberg S (2002) Competitive adsorption of arsenate and arsenite on oxides and clay minerals. Soil Sci Soc Am J 66:413–421
Goodbred SL Jr, Kuehl SA (2000) The significance of large sediment supply, active tectonism, and eustasy on margin sequence development: late quaternary stratigraphy and evolution of the Ganges–Brahmaputra delta. Sediment Geol 133:227–248
Grasshoff K, Ehrhardt M, Kremling K (1983) Methods of seawater analysis, 2nd edn. Verlag Chemic, Germany
Grossl PR, Eick M, Sparks DL, Goldberg S, Ainsworth CC (1997) Arsenate and chromate retention mechanisms on goethite. 2. Kinetic evaluation using a pressure–jump relaxation technique. Environ Sci Technol 31:321–326
Hering JG, Kneebone PE (2002) Biogeochemical controls on arsenic occurrence and mobility in water supplies. In: Frankenberger WT Jr (ed) Environmental chemistry of Arsenic. Marcel Dekker, New York, p 161
Jones CA, Langner HW, Anderson K, McDermott TR, Inskeep WP (2000) Rates of microbially mediated arsenate reduction and solubilization. Soil Sci Soc Am J 64:600–608
Kirk MF, Holm TR, Park J, Jin Q, Sanford RA, Fouke BW, Bethke CM (2004) Bacterial sulfate reduction limits natural arsenic contamination in groundwater. Geol 32(11):953–956. doi:10.1130/G20842.1
Kumar SR (1996) Distribution of organic carbon in the sediments of Cochin mangroves, southwest coast of India. Indian J Mar Sci 25:274–276
Langmuir D, Mahoney J, Rowson J (2006) Solubility products of amorphous ferric arsenate and crystalline scorodite (FeAsO4.2H2O) and their application to arsenic behavior in buried mine tailings. Geochim Cosmochim Acta 70:2942–2956
Lee SZ, Allen HE, Huang CP, Sparks DL, Sanders PF, Peijnenburg WJGM (1996) Predicting soil–water partition coefficients for cadmium. Environ Sci Technol 30(12):3418–3424
Loring DH, Rantala RTT (1992) Manual for geochemical analysis of marine sediment and suspended particulate matter. Earth Sci Rev 32:235–283
Lowers HA, Breit GN, Foster AL, Whitney J, Yount J, Uddin MdN, Muneem A (2007) Arsenic incorporation into authigenic pyrite, Bengal Basin sediment, Bangladesh. Geochim Cosmochim Acta 71:2699–2717
Lu Y, Pignatello JJ (2002) Demonstration of the “conditioning effect” in soil organic matter in support of a pore deformation mechanism for sorption hysteresis. Environ Sci Technol 36:4553–4561
Manceau A (1995) The mechanism of anion adsorption on iron oxides: evidence for the bonding of arsenate tetrahedra on free Fe (O, OH)6 edges. Geochim Cosmochim Acta 59(17):3647–3653
Mandal SK, Dey M, Ganguly D, Sen S, Jana TK (2009) Biogeochemical controls of arsenic occurrence and mobility in the Indian Sundarban mangrove ecosystem. Mar Pollut Bull 58(5):652–657. doi:10.1016/j.marpolbul.2009.01.010
Meharg AA, Scrimgeour C, Hossain SA, Fuller K, Cruickshank K, Williams PN, Kinniburgh DG (2006) Codeposition of organic carbon and arsenic in Bengal Delta aquifers. Environ Sci Technol 40(16):4928–4935
Mertal J, Charlet L, Burean S, Mallik SB, Chakraborty S, Ahmed KM, Rahman MW, Cheng Z, Vangeen A (2008) Comparison of dissolved and particulate arsenic distribution in shallow aquifers of Chakdaha, India and Araihazar, Bangladesh. Geochem Trans 9:1. doi:10.1186/1467-4866-9-1
Mott CJB (1981) Anion and ligand exchange. In: Green DJ, Hays MHB (eds) The Chemistry of soil processes. Wiley Interscience, New York
National Institute of Standards and Technology (2003) Critically selected stability constants of metal complexes database. version 7.0. U.S. Department of Commerce, Gainsburg
Pan G, Liss PS, Krom MD (1999) Particle concentration effect and adsorption reversibility. Colloids Surf A Physicochem Eng Asp 151:127–133
Parfitt RL, Atkinson RJ, Smart RSC (1975) The mechanism of phosphate fixation by iron oxides. Soil Sci Soc Am Proc 39:837–841
Ray R, Ganguly D, Chowdhury C, Dey M, Das S, Dutta MK, Mandal SK, Majumdar N, De TK, Mukhopadhyay SK, Jana TK (2011) Carbon sequestration and annual increase of carbon stock in a mangrove forest. Atmos. Env. doi:10.1016/j.atmosenv.2011.04.074
Sarkar SK, Franciskovic-Bilinski S, Bhattacharya AK, Saha M, Bilinski H (2004) Levels of elements in the surficial estuarine sediments of the Hugli River, northeast India and their environmental implications. Environ Int 30:1089–1098
Shrawat KL (1982) Simple modification of the Walkley–Black method for simultaneous determination of organic carbon and potentially mineralizable nitrogen in tropical rice soils. Plant Sci 69:73–77
Siegel M, Aragon A, Zhao H, Deng S, Nocon M, Aragon M (2008) Prediction of arsenic removal by adsorptive media. In: Comparison of field and laboratory studies in arsenic contamination of groundwater. John Wiley & Sons, Inc.227
Silva Filho EV, Jonathan MP. Chatterjee M, Sarkar SK, Sella SM, Bhattacharya A, Satpathy KK (2010) Ecological consideration of trace element contamination in sediment cores from Sundarban Wetland, India, Environ Earth Sci. doi:10.1007/s 12665-010-079S-9
Sims JT, Heckendorn SE (1991) Methods of analysis of the university of delaware soil testing laboratory. Cooperative bulletin no. 10, University of Delaware, Newark
Sposito G (1984) The surface chemistry of soils. Oxford University Press, New York
Villapando RR, Graetz DA (2001) Phosphorus sorption and desorption properties of the spodic horizon from selected Florida Spodosols. Soil Sci Soc Am J 65:331–339
Vischer PT, Beukema J, van Gemerden H (1991) In situ characterization of sediments: measurements of oxygen and sulfide profiles with a novel combined needle electrode. Limnol Oceanogr 36:1476–1480
Wagemann R (1978) Some theoretical aspects of stability and solubility of inorganic arsenic in the fresh water environment. Water Res 12:139–145
Wauchope RD, McDowell LL (1984) Adsorption of phosphate, arsenate, methanearsonate and cacodylate by lake and stream sediments: comparisons with soils. J Environ Qual 13:499–504
Webster JG (1990) The solubility of As2S3 and speciation of As in dilute and sulphide bearing fluids at 25 and 90° C. Geochim Cosmochim Acta 54(4):1009–1017
Wu J, Laird DW, Thompson ML (1999) Sorption and desorption of copper on soil clay components. J Environ Qual 28:334–338
Xu HB, Allard B, Grimvall A (1988) Influence of pH and organic substance on the adsorption of As(V) on geologic materials. Water Air Soil Pollut 40:293–305
Yamamoto M, Yasuda M, Yamamoto Y (1985) Hydride generation atomic absorption spectroscopy coupled with flow injection analysis. Anal Chem 57:1375–1382
Zobrist J, Dowdle PR, Davis JA, Oremland RS (2000) Mobilization of arsenite by dissimilatory reduction of adsorbed arsenate. Environ Sci Technol 34:4747–4753
Acknowledgments
One of the authors (N.M.) is grateful to UGC, New Delhi for providing fellowship. Thanks are also due to the Sundarban Biosphere Reserve and Divisional Forest Office, Govt. of West Bengal, for giving permission to carry out the experiments. We are grateful to the peer reviewers for their helpful and constructive suggestions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mandal, S.K., Majumder, N., Chowdhury, C. et al. Adsorption kinetic control of As(III & V) mobilization and sequestration by Mangrove sediment. Environ Earth Sci 65, 2027–2036 (2012). https://doi.org/10.1007/s12665-011-1183-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12665-011-1183-9