
Abstract
The receptor for advanced glycation end products (RAGE) at the blood-brain barrier (BBB) is critical for regulation of amyloid-β (Aβ) homeostasis in the diabetic brain. In this study, we used an in vitro BBB model consisting of mouse brain capillary endothelial cells (MBCECs) to investigate whether advanced glycation end products (AGEs) increase Aβ influx transport across the BBB and the underlying mechanisms. We found that AGEs induced Aβ influx transport across the BBB in concentration- and time-dependent manner, accompanied by increased RAGE expression and nuclear factor-kappa B p65 (NF-κB p65), and decreased nuclear peroxisome proliferator-activated receptor γ (PPARγ). Blockade of RAGE with its antibody and inhibition of NF-κB signaling with PDTC as well as activation of PPARγ with rosiglitazone significantly decreased Aβ transport across the BBB from the periphery to the brain. These treatments also pronouncedly suppressed AGEs-induced increases in RAGE expression and nuclear NF-κB p65 and reversed the decrease in nuclear PPARγ. These results suggest that RAGE-NF-κB-PPARγ signaling is involved in regulation of AGEs-induced influx transport of Aβ across the BBB and targeting the signaling pathway could serve as a novel strategy to modify such Aβ transport.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A growing number of clinical and epidemiological evidences correlate a strong connection between type 2 diabetes (T2D) and Alzheimer’s disease (AD) (Arvanitakis et al. 2004; Sanz et al. 2012; Stewart and Liolitsa 1999). Several pathological indications such as disrupted glucose metabolism and insulin uptake, and cognitive decline lie under the same bridge between T2D and AD (Arvanitakis et al. 2004; Liu et al. 2011a, b). Moreover, dysfunctional brain insulin signaling has been reported in the pathogenesis of AD (Biessels and Reagan 2015; Hoyer and Nitsch 1989). Recent studies suggest that diabetes may alter blood-brain barrier (BBB) functioning, owing to an abnormal transportation of molecules through it, which may contribute to AD-related cognitive impairment (Hong et al. 2009; Huber 2008; Liu et al. 2009; Neeper et al. 1992). The BBB is a monolayer of brain endothelial cells, tightly sealed with paracellular protein complexes with limited intercellular spaces and connected at a junctional complex by the tight junctions and the adherens junctions. The BBB is bound by an extracellular matrix, and maintained through interactions between pericytes and glial cells (Zlokovic 2008). It is critical for maintaining brain homeostasis and brain functioning by means of exchanging materials between the periphery and the central nervous system (CNS). Exchange of a majority of molecules, except for the molecules with a smaller molecular weight than 400 Da (Pardridge 2007), through the BBB is impermissible (Ohtsuki and Terasaki 2007), a mechanism by which pathogenic entry into the brain is also safeguarded. The scaffold proteins at the tight junctions of the BBB are mainly affected during a pathogenic exposure. Dysfunction of the BBB with efflux and influx transporters has also been reported in the pathogenesis of several neurodegenerative disorders (Ueno et al. 2010). Advanced glycation end products (AGEs) and the receptor for AGEs (RAGE) have been implicated in a range of pathological circumstances such as in diabetes to cardiovascular disease, renal disease to neurological diseases. Diabetes and its related pathologies such as diabetic neuropathy, diabetic nephropathy (Youssef et al. 1999) and diabetes-related cardiovascular complications have been strongly correlated with RAGE. RAGE, a member of the multiligand cell surface immunoglobin family that exists at the lumina of the endothelial cytoplasm of the BBB, is involved in the receptor mediated amyloid-β (Aβ) trafficking across the BBB (Deane et al. 2004a, 2004b; Wan et al. 2015; Zhang and Lee 2011), and is critical for maintaining Aβ homeostasis in the brain (Deane et al. 2003; Shibata et al. 2000). The role of the BBB in the pathogenesis of AD has also been shown in human specimens with a positive correlation with RAGE expression (Provias and Jeynes 2014). Our recent studies showed a significant increase in RAGE-dependent Aβ influx across the BBB, accompanied by cerebral Aβ accumulation and memory impairment in diabetic mice with hypoinsulinemia (Liu et al. 2009, 2013) and hyperinsulinemia (Chen et al. 2016). Herein, we used a monolayer of in vitro BBB model to investigate the changes in Aβ transport across the BBB and the restorative effects of rosiglitazone, a thiazolidinedione group of anti-diabetic drug that act by selectively agonizing the effects at PPARγ receptors on the adipocytes; we also used PDTC (pyrrolidinedithiocarbamate) that can reversibly inhibit the activation of IκB-NF-κB interaction that is critical for NF-κB mediated gene expressions (Schreck et al. 1992); and used antibodies against RAGE to knockdown its effects on mediation of abnormal transport of Aβ across the BBB.
Materials and Methods
Generation of AGEs in BSA
Solution of AGEs in bovine serum albumin (BSA, NanJing SunShine Biotechnology Co., LTD., China) was prepared as previously described with minor modifications (Liu et al. 2011a, 2011b). Briefly, 50 mg/ml BSA was incubated under sterile conditions with 0.5 M D-glucose (Sinopharm Chemical Reagent Co., LTD., China) in 0.1 M phosphate buffered saline (PBS, pH 7.4) at 37 °C for 10 weeks in a dark place. Unincorporated glucose was removed by dialysis against PBS. Control BSA was incubated under the same conditions, in absence of the reducing glucose. Protein concentrations were determined by the Bradford method.
Estimation of AGEs Formation
Characterization of AGEs was based on lysine residue modifications and their fluorescent properties. The fraction of modified lysine residues was measured by 2,4,6-trinitrobenze-nesulfonic acid method (TNBS, Sigma-Aldrich, USA) that estimates the proportion of unmodified lysine in AGEs preparation compared with that of the unmodified BSA (Shi et al. 2013). As a result, the extent of lysine modification was 87% compared to unmodified BSA (Fig. 1a). Extent of fluorescent AGEs formation was assessed by spectrofluorometry. AGEs and unmodified BSA was diluted with PBS, and fluorescent intensity was recorded at excitation 360 nm, emission 440 nm. The characteristic glycation fluorescence of AGEs was increased by approximately 12-fold compared with unmodified BSA. This indeed strongly suggested that AGEs have been formed (Fig. 1b, c).

AGEs are characterized based on lysine residue modifications and their fluorescence properties. (a) The fraction of modified lysine residues was measured by TNBS method that estimated the proportion of unmodified lysine in AGEs preparation compared with that of the unmodified BSA. Data are given as means ± SEM, n = 6. AGEs (b) and unmodified BSA (c) were diluted with PBS, and fluorescence intensity was recorded at excitation 360 nm, emission 440 nm
Isolation and Culture of Primary Mouse Brain Capillary Endothelial Cells (MBCECs)
Endothelial cells from brain capillaries were isolated from 12-week-old mice (Yangzhou University Medical Center, China) and cultured according to previously reported methods with some modifications (Hong et al. 2006; Nishitsuji et al. 2011; Wuest and Lee 2012). Briefly, mice were euthanized according to local IACUC/veterinarian recommendations, fresh brains were collected immediately and placed in an ice-cold Hank’s Balanced Salt Solution, and the meninges and superficial blood vessels were removed. Then the gray matter was collected, triturated and digested for 2 h at 37 °C in a solution containing 1 mg/ml of type II collagenase (NanJing SunShine Biotechnology Co., LTD., China) and 28.3 U/mL DNase I (NanJing SunShine Biotechnology Co., LTD., China) in ice-cold Dulbecco’s modified Eagle’s medium (DMEM, Hyclone, USA). To remove myelin, the digest in 20% BSA in DMEM was centrifuged at 1000g for 20 min to obtain cell pellets. The microvessels obtained in the pellets were digested further with 1 mg/ml of collagenase/dispase (Roche, Germany) in DMEM for 1.5 h at 37 °C. The digested microvessel solution was centrifuged at 700g, 4 °C for 6 min. The pellets were resuspended and layered over a 33% continuous Percoll gradient (NanJing SunShine Biotechnology Co., LTD.) and centrifuged at 1000g for 10 min at 4 °C. Subsequently, the layer containing microvessels was removed and diluted in DMEM. After centrifugation at 700g for 10 min, cell pellets were resuspended and used for cultivation. Cells were washed with DMEM and resuspended in culture media consisting of DMEM supplemented with 20% fetal bovine serum (Gibco-Invitrogen, USA), 2 mM L-glutamine (Sigma-Aldrich, USA), 1.5 ng/ml basic fibroblast growth factor (Pepro Tech, USA), 100 μg/ml heparin (Sigma-Aldrich, USA), an antibiotic solution (100 U/mL penicillin, 0.25 μg/mL Gentamycin Sulfate) and 4 g/ml puromycin (Sigma-Aldrich, USA). The culture medium was changed on day 2. After day 2, puromycin was omitted and medium was changed every 3 days. Cell suspension from one brain was seeded per well of a 12-well plate. Experiments were performed when cells reached confluence, usually after 1 week in culture at 37 °C and 5% CO2.
Immunocytochemical Identification of MBCECs
Previous studies have shown that the purity of MBCECs isolated by the present method, based on immunocytochemical detection of the expression of factor VIII-related antigen (von Willebrand factor), is more than 95% (Chen et al. 2007). To verify if the isolated brain cells were MBCECs in our preparation, immunocytochemical analyses of factor VIII were carried out following the standard protocol provided with the SABC kit (Boster Biotechnology Co., Ltd., China). MBCECs were plated on glass slides in one well of a 24-well plate (Corning, USA) with 600 μl of culture medium unless 80% confluency was reached. Slides were fixed in 4% paraformaldehyde solution for 30 min, and blocked with PBS containing 3% normal goat serum, 0.3% (v/v) Triton X-100, and 0.1% BSA at room temperature for 30 min. Cells were incubated with a rabbit polyclonal antibody against factor VIII protein (Bioworld Technology Co., Ltd., China), which was diluted 1:100 in blocking buffer, overnight at 22 °C in a humidified chamber. Negative controls were prepared without the primary antibody in a humidified chamber. Then cells were washed in 0.01 M PBS (6 × 5 min, 22 °C), incubated with a mouse IgG antibody conjugated with biotin (1:200 in 0.01 M PBS; 1 h, 22 °C), washed in 0.01 M PBS (3 × 5 min, 22 °C), and then incubated with streptavidin-biotin complex (SABC) (1:200 in 0.01 M PBS; 20 min, 22 °C). After a further 3 × 5 min washes in 0.01 M PBS, DAB was used as the final chromogenat 22 °C for detection of factor VIII. Photomicrographs were obtained using a microscope (Olympus DP72, Japan).
The Transendothelial Electrical Resistance (TEER)
MBCECs were seeded (200,000 cells/cm2) onto 2% gelatin (Sigma-Aldrich, USA)-coated transparent and porous membrane of cell culture insert. After 3–5 days, cultured in a 5% CO2–95% air incubator at 37 °C, confluent monolayer of MBCECs was generated (Xie et al. 2005). The culture medium was removed and then washed three times with PBS. After the insert was dropped into PBS, the barrier function of the endothelial monolayer was estimated by measuring the transendothelial electrical resistance (TEER) with a Millicell ERS ohmmeter (Millipore, USA), as previously reported (Chen et al. 2014). The values were corrected for the background resistance measured across the filter without cells.
Measurement of Monolayer MBCECs Permeability
The permeability of MBCECs was assessed by the passage of FITC-conjugated dextran (MW 40000; Sigma-Aldrich) through a monolayer cell, as previously described with some modifications (Fang et al. 2016). In brief, MBCECs were grown to confluence on transwell filter inserts (24-well format, 0.4 mm pore; Corning, USA). Culture medium with 1 mg/ml FITC-dextran was added into the upper chamber. A total of 1 ml culture medium without FITC-dextran was added to the lower chamber. Samples (50 μl) were taken from lower chamber at 1 h, and analyzed by a fluorescence microplate reader (PerkinElmer, Waltham, USA) according to λex488 nm and λem515 nm. A permeability coefficient (Pe, cm/s) was determined, where permeability coefficient = V/(A s × C d) × (C r/T), where V is the medium volume in receiver chamber, A s is the surface area of cell monolayer, C d is the concentration of FITC-conjugated dextran in the donor chamber at time 0, and C r is the concentration of FITC-conjugated dextran in the receiver chamber at sampling time T. All data were from at least 3 independent experiments.
AGEs and Drug Treatment in Cell Culture
MBCECs were rinsed with PBS, replenished with fresh serum-free media, and cultured for an additional 24 h. To evaluate the effect of AGEs on RAGE expression in MBCECs, the cells were incubated with 200 mg/ml of AGEs for 6, 12 and 24 h or treated with 50, 100 and 200 mg/ml of AGEs for 24 h. Moreover, we observed the effect of anti-RAGE immunoglobulin G (5 μg/ml, Abcam Technology Co., Ltd., China), preimmune IgG (5 μg/ml, R&D Systems, Inc., China), PDTC (10 μM, Sigma-Aldrich, USA) and rosiglitazone (Rsg, 1 μM, Sigma-Aldrich, USA) on the level of RAGE in MBCECs and explored whether they could regulate the effect of AGEs.
Measurement of [125I]-Amyloid-β1–40 (Aβ1–40) Kinetics
[14C]-inulin was purchased from American Radiolabeled Chemicals, Inc. Aβ1–40 was from Sigma-Aldrich. Na125I was purchased from Chengdu GaoTong Isotope Co., Ltd. Human [125I]-Aβ1–40 were synthesized at the Jiangsu Institute of Nuclear Medicine. To analyze the integrity of [125I]-Aβ1–40 over a 6 h of time period, 0.1 nM [125I]-Aβ1–40 was incubated in serum-free medium at 37 °C before samples were subjected to trichloroacetic acid (TCA) precipitation. 50 μL of 15% TCA was added to a 50 μL media sample and incubated for 10 min at 4 °C. Samples were then centrifuged at 10,000g for 10 min. Supernatant (free [125I]) and pellet (intact [125I]-Aβ1–40) were counted separately for [125I] on a 1470 automatic gamma counter (PerkinElmer, USA). Results were expressed as mean ± SEM of 3 independent experiments.
In Vitro Transwell MBCECs Model for Transport Studies
Our transwell transport model set up resembles a previously published method with minor modifications (Pflanzner et al. 2014). In brief, freshly isolated MBCECs fragments were seeded onto coated transwell filters with membrane pore size 0.4 μm and cultured as described above. After incubation for 24 h in serum-free medium, MBCECs were pretreated with anti-RAGE antibody, PDTC or Rsg for 2 h, followed by AGEs (200 μg/ml) for 24 h, and then the treated cells were used for transport studies. [125I]-Aβ1–40 (0.1 nM) and [14C]-inulin (1 μCi/mL), a marker for paracellular diffusion, were added to serum-free media and incubated at 37 °C. From each input and at each time point, 10 and 50 μL samples were taken from the compartment the transport was investigated to (defined as acceptor compartment). 10 μl probes were counted on a 1470 automatic gamma counter or [125I], or on a Tri-Carb 2800 TR Liquid Scintillation Analyzer (PerkinElmer, USA) for [14C]. To investigate the amount of intact [125I]-Aβ1–40 being transported, 50 μL 15% TCA was added to a 50-μL media sample and incubated for 10 min at 4 °C. Samples were then centrifuged at 10,000g for 10 min. Supernatant (free [125I]) and pellet (intact [125I]-Aβ1–40) were counted separately for [125I]. Transport of intact [125I]-Aβ1–40 across the monolayer was calculated as Aβ1–40 transcytosis quotient (TQ):
For 6 h time course transport studies, MBCECs were cultured on 24-well transwell inserts (growth surface area 33 mm2) with 600 μL media in the upper (luminal) compartment and 1.8 mL in the lower (abluminal) compartment, resembling blood vessel and brain, respectively. [125I]-Aβ1–40 and [14C]-inulin were added to the luminal (input) compartment and samples were taken at each time point from the abluminal (acceptor) compartment. Results represent mean ± SEM of 3 independent experiments.
Western Blot Analysis
MBCECs were washed with PBS and lysed in NP-40 buffer containing proteinase inhibitors (Roche, Germany). Protein concentration was determined by the BCA protein assay kits reagent (Pierce, USA) assessed for expression of RAGE. Protein extracts were separated by a SDS-polyacrylamide gel electrophoresis and then transferred onto a PVDF membrane. The membrane was blocked with 5% skim milk in Tris buffer saline and then incubated at 4 °C overnight with respective primary antibodies for rat anti-RAGE antibody (1:500), or β-actin (inner control, 1:3000). After washing with TBST, the membranes were incubated with a horseradish peroxidase conjugated secondary antibody (1∶5000) for 2 h at room temperature. The antibody-reactive bands were visualized by using the enhanced chemiluminescence detection reagents by a gel imaging system (Tanon Science & Technology Co., Ltd., China).
Nuclear extracts were prepared using nucleoprotein extraction kit (Sangon Biotech, China). Briefly, cell monolayers were washed twice with cold PBS and scraped into cold PBS (1.0 ml) and nuclear extracts were prepared following the standard protocol provided by nucleoprotein extraction kit. Briefly, the cell suspension was centrifuged for 10 min at 1000g at 4 °C. The cell pellet was then resuspended in 0.4 ml of ice-cold hypotonic buffer containing 0.5% phosphatase inhibitor, 1% phenylmethylsulfonyl fluoride and 0.1% DL-dithiothreitol incubated on ice for 15 min. After vortexing for 15 s, cell lysates were centrifuged for 5 min at 3000g at 4 °C, the supernatant was removed, and the nuclear pellet was washed once with hypotonic buffer and centrifuged at 4 °C, 5000g for 5 min. Finally, 0.2 ml lysis buffer containing 0.5% phosphatase inhibitor, 1% phenylmethylsulfonyl fluoride and 0.1% DL-dithiothreitol were added with the precipitate, chilled for 20 min and centrifuged at 4 °C, 15,000g for 10 min. The supernatant nuclear protein extract was subjected to Western blot for assay of NF-κB p65, PPARγ and histone H3 was used as a loading control. Antibodies were purchased from different companies: rabbit anti-mouse NF-κB p65 and PPARγ from Cell Signaling Technology, Inc. (USA); rabbit anti-mouse Histone H3, β-actin and goat anti-rabbit secondary antibodies from Bioworld Technology Co., Ltd. (USA).
Coimmunoprecipitation
Coimmunoprecipitation was performed as described previously (Yin et al., 2002). A total of 2 × 107 MBCECs were pelleted and then lysed in lysis buffer (1% Triton X-100, 150 mM NaCl, 10 mM Tris, pH 7.4, 1 mM EDTA, 1 mM EGTA, pH 8.0, 0.2 mM sodium orthovanadate, 0.5% NP-40, 0.2 mM PMSF, 4 g/ml pepstatin, 4 g/ml leupeptin, 5 g/ml aprotinin) on ice for 30 min. Cellular debris was removed by centrifugation at 16,000g for 20 min, and the supernatant was incubated with rabbit anti-mouse NF-κB p65 at a concentration of 4 μg/ml at 4 °C for 3 h. Immunocomplex was captured by incubating the samples with 50 μl protein A + G–agarose (Beyotime, China) suspension overnight at 4 °C. Agarose beads were collected by centrifugation and washing. After microcentrifugation, the pellet was resuspended in SDS loading buffer, separated on an SDS-polyacrylamide gel, transferred to PVDF membrane, and further analyzed by Western blot using rabbit anti-PPARγ antibody as described previously. Affinity-purified normal rabbit IgG (Santa Cruz Biotechnology, USA) was used as a negative control for immunoprecipitation.
Semi-Quantitative Reverse Transcription-Polymerase Chain Reaction (RT-PCR) Assay
Total RNA was extracted from MBCECs using Trizol reagents according to the manufacturer’s instructions. For cDNA synthesis, aliquots of total RNA (2 μg) were mixed with 0.2 μg random hexamer primer, 20 U RNasin, 1 mM dNTP, and 200 UM-MuLV reverse transcriptase in 20 μl of the reverse reaction buffer. The mixture was incubated at 25 °C for 10 min, 42 °C for 60 min and then at 72 °C for 10 min to deactivate the reverse transcriptase. PCR was performed on an Eppendorf Master Cycler (Eppendorf, Germany). The mixture contained: 1 μl RT-cDNA template dissolved in 20 μl reaction mixture containing 1 × PCR buffer, 200 μM dNTP,1.5 mM MgCl2, 20 pmol of each primer and 0.5 U Taq DNA polymerase. Cycling parameters were as below: 94 °C for 1 min, followed by 33 cycles of 94 °C for 30 s, 63 °C for 30 s and 72 °C for 1 min, with a final extension step of 72 °C for 7 min. The abundance of transcripts in cDNA samples was measured by RT-PCR with the following primers: mouse RAGE forward 5′-ACATGTGTGTCTGAGGGAAGC-3′ and reverse 5′-AGCTCTGACCGCAGTGTAAAG-3′ (254 bp, nucleotides 956–1169 in L33412, GeneBank, Invitrogen), β-actin forward 5′-TCTTGGGTATGGAATCCTGTG-3′ and reverse 5′-ATCTCCTTCTGCATCCTGTCA-3′ (154 bp, nucleotides 876–1029 in NM_007393.3, GeneBank, Invitrogen). The amplification products were separated by electrophoresis on a 2% agarose gel containing ethidium bromide. The optical density of the bands was determined by an image analysis system (Tanon Science &Technology Co. Ltd., China).
Statistical Analyses
Data shown were expressed as mean ± S.E.M. All data, unless specified, were analyzed by a one-way ANOVA followed by a Dunnett’s post hoc analysis for multiple comparisons. At each time point transport studies were analyzed using a one-way ANOVA followed a Turkey’s post hoc analysis. All analyses were carried out using SPSS, version 20.0. The level of statistical significance was P < 0.05.
Results
Characterization of the MBCECs Culture
The isolated microvessels, prepared according to the procedure described, were pure, without any obvious growth of neuronal or glial cells. After digestion, the endothelial cells displayed the appearance of “beads on a string” (Fig. 2a), then MBCECs started to migrate out from the vessels on day 2 and grew to confluency within 7–10 days. Primary endothelium culture displayed typical “cobble stone” morphology (Fig. 2b). Based on morphology, >95% of the population consisted of endothelial cells. Moreover, positive immunostaining for von Willebrand factor, a marker for endothelial cells, was found to be present diffusely in the cytoplasm of cultured MBCECs (Fig. 2c, e). Negative control slides remained unstained (Fig. 2d, f). We also estimated the barrier function of the endothelial monolayer by measuring TEER. Culture with serum-free medium for 24 h induced higher TEER (Fig. 2g). TEER increased in a time-dependent manner after preparing the in vitro BBB model, but it decreased after more than 24 days (Fig. 2g). Permeability studies, to supplementally detect the function of BBB in vitro, were performed to check the tight junctions integrity by using FITC-conjugated dextran. The endothelial permeability coefficient (Pe) for dextran of in vitro BBB model is as low as 3 × 10−6 cm s−1. Hence, we used MBCECs cultured for 24 days (passage 3) for further experiments.

Primary culture of MBCECs and identification of BBB in vitro. a “Beads on string” microvessels after digestion (100×). b “Pebble” like MBCECs after passage (100×). (c, e) MBCECs at passage 3 cultured on gelatin-coated chamber slides stained positively with antibody to VIII factor (c, 100×; e, 200×). (d, f) Negative control slide remained unstained (d, 100×; f, 200×). g After MBCECs were cultured at day 10 (primary culture), day 17 (passage 2), day 24 (passage 3), day 31 (passage 4), and day 38 (passage 5), TEER was recorded under different conditions. Culture with serum-free medium for 24 h induced higher TEER. Data are given as means ± SEM, n = 6 (CM complete medium, SFM serum-free medium)
AGEs Increase RAGE Expression in MBCECs
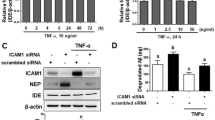
MBCECs were incubated with 200 mg/ml of AGEs for 6, 12 and 24 h or treated with 50, 100 and 200 mg/ml of AGEs for 24 h. As shown in Fig. 2, MBCECs treated with 200 mg/ml of AGEs for 6 h (P = 0.394, Fig. 3a, b) or with 50 mg/ml of AGEs for 24 h (P = 0.382, Fig. 3c, d) did not affect RAGE expression. Stimulation of MBCECs with AGEs (200 mg/ml) for 24 h led to a 2.5-fold increase in cell surface RAGE expression (P < 0.01, Fig. 3a, b). Exposure of MBCECs to AGEs induced a time-dependent (F(4,15) = 9.633, P < 0.05, Fig. 3a, b) and dose-dependent (F(4,15) = 8.360, P < 0.05, Fig. 3c, d) increase in the level of RAGE protein.

AGEs increase RAGE expression in MBCECs. The protein level of RAGE was detected by WB and β-actin was used as a loading control. a MBCECs were incubated with AGEs (200 mg/ml) for 6, 12, and 24 h before whole-cell lysates were extracted and then subjected to WB. c MBCECs were treated with 50, 100, and 200 mg/ml of AGEs for 24 h. Quantification of RAGE (b, d) was expressed as the ratio (in percentage) of Veh + Veh. Values shown are expressed as mean ± SEM; n = 4. *P < 0.05, **P < 0.01 vs Veh + Veh group
Integrity of [125I]-Aβ1–40
Before the commencement of the transcytosis experiment, we determined the stability of [125I]-Aβ1–40 under similar conditions used during the transcytosis experiment. TCA precipitation of media samples enabled us to differentiate between intact [125I]-Aβ1–40 (TCA-precipitable) and free [125I] (non TCA-precipitable). The experiment revealed a minor disintegration of [125I] from [125I]-Aβ1–40 during the 6 h time course as shown by the decrease in [125I]-Aβ1–40 counts (F(3,28) = 1.620, P = 0.260; Fig. 4a) and the concomitant increase in [125I] counts (F(3,28) = 1.734, P = 0.237; Fig. 4b). Hence, in further experiments we only considered TCA precipitated intact [125I]-Aβ1–40 to calculate the Aβ1–40 transcytosis quotient (TQ).

Only a small amount of [125I] spontaneously disintegrates from [125I]-Aβ1–40 within 6 h incubation. [125I]-Aβ1–40 (0.1 nM) was incubated in medium at 37 °C before samples were subjected to TCA precipitation. Intact [125I]-Aβ1–40 and free [125I] counts per minute (CPM) were analyzed separately on a γ-counter. A minor disintegration of [125I] from [125I]-Aβ1–40 during the 6 h time course is shown by the decrease in [125I]-Aβ1–40 CPM (a) and the concomitant increase in [125I] CPM (b). Results are expressed as mean ± SEM of 8 independent experiments
AGEs Increase Aβ Influx Transport Across the BBB In Vitro (Transcytosis Measurement)
In a first set of experiments, we investigated the transport of [125I]-Aβ1–40 from the luminal to abluminal direction (blood-to-brain) across MBCECs monolayer cultured in 24-well transwell dishes over a 6-h time period. AGEs enhanced transport of iodinated Aβ1–40 influx transport across BBB in vitro at each time point (F(2,9) = 7.370, P < 0.05, Fig. 5 for 2 h; F(2,9) = 11.345, P < 0.05, Fig. 5 for 4 h; F(2,9) = 9.261, P < 0.05, Fig. 5 for 6 h). After TCA precipitation of media samples, we were able to distinguish intact [125I]-Aβ1–40 (TCA-precipitable) from free [125I] (non TCA-precipitable, generated during [125I]-Aβ1–40 degradation by MBCECs or spontaneous [125I]-Aβ1–40 disintegration).

AGEs induce an increase in Aβ influx transport across BBB in vitro. MBCECs were cultured on gelatin-coated transwell inserts for 5 days before high transendothelial electrical resistance was induced for 24 h by serum withdrawal. Subsequently, transport studies for [125I]-Aβ1–40 were conducted in the presence of [14C]-inulin as a marker for paracellular diffusion. AGEs increased the [125I]-Aβ1–40 transport from luminal to abluminal compartment at each time point compared with control. Results are expressed as mean ± SEM, n = 4 per group. *P < 0.05 vs Veh + Veh group
Anti-RAGE Antibody, PDTC and Rosiglitazone Reverse AGEs-Induced Upregulation of Aβ Influx Transport Across the BBB
Treatment with anti-RAGE antibody (5 μg/ml), PDTC (10 μM) and rosiglitazone (1 μM) effectively attenuated AGEs-upregulated apical-to-basolateral passage of [125I]-Aβ1–40 across the BBB (Fig. 5). At each time point of transcytosis experiment, anti-RAGE antibody restored AGEs-upregulated transport of [125I]-Aβ1–40 from luminal compartment into abluminal compartment (F(6,21) = 5.637, P < 0.05, Fig. 6a for 2 h; F(6,21) = 5.215, P < 0.05, Fig. 6a for 4 h; F(6,21) = 4.568, P < 0.05, Fig. 6a for 6 h), however, IgG had no effect on influx transport of [125I]-Aβ1–40 across the BBB model in vitro. PDTC reduced the amount of [125I]-Aβ1–40 in abluminal compartment by approximately 30% compared with Veh + AGEs group at the last point (F(4,15) = 7.793, P < 0.05, Fig. 6b for 4 h; F(4,15) = 7.662, P < 0.05, Fig. 6b for 6 h). Further, we detected a marked difference in the abluminal acceptor compartment for rosiglitazone group compared with Veh + AGEs group from 4 h, which indicated rosiglitazone significantly decreased AGEs-induced upregulation of Aβ influx transport across the BBB (F(5,18) = 8.201, P < 0.05, Fig. 6c for 4 h; F(5,18) = 7.825, P < 0.05, Fig. 6c for 6 h).

Anti-RAGE antibody, PDTC and rosiglitazone (Rsg) reverse AGEs-induced upregulation of Aβ influx transport across BBB in vitro. (a, b, c) Pretreatment of MBCECs with anti-RAGE antibody (5 μg/ml), PDTC (10 μM) or rosiglitazone (1 μM) for 2 h decrease [125I]-Aβ1–40transport from luminal to abluminal compartment compared with MBCECs exposed to AGEs alone. Results represent mean ± SEM of 4 independent experiments. *P < 0.05 vs AGEs + Veh group
Anti-RAGE Antibody, PDTC and Rosiglitazone Reverse AGEs-Upregulated RAGE Expression
Our transport experiment suggested anti-RAGE antibody, PDTC and rosiglitazone decrease AGEs-increased transcytosis of [125I]-Aβ1–40 from blood to brain. It has been demonstrated that RAGE is a main protein involved in receptor-mediated Aβ influx across the BBB. Then we detected RAGE protein in MBCECs treated or untreated with anti-RAGE antibody, PDTC, rosiglitazone by Western blot. Interestingly, protein expression of RAGE was significantly reduced in MBCECs pretreated with anti-RAGE antibody, PDTC and rosiglitazone (F(6,21) = 4.971, P < 0.05, Fig. 7a, b; (F(4,15) = 5.085, P < 0.05, Fig. 7c, d; F(5,18) = 6.459, P < 0.05, Fig. 7e, f). RT-PCR analysis further revealed that anti-RAGE antibody, PDTC and rosiglitazone decreased AGEs-upregulated RAGE protein via inhibition of its mRNA transcription (data not shown).

Anti-RAGE antibody, PDTC and rosiglitazone decrease AGEs-induced upregulation of RAGE expression. MBCECs were treated with anti-RAGE antibody, PDTC or rosiglitazone, followed by incubation with AGEs for 24 h. Representative immunoblot of RAGE detected by Western blot (a, c, e), protein quantification was expressed as the ratio (in percentage) of Veh + Veh group (b, d, f). Values shown are expressed as mean ± SEM; n = 3–4. *P < 0.05, **P < 0.01 vs AGEs + Veh group
Anti-RAGE Antibody, PDTC and Rosiglitazone Inhibit AGEs-Induced NF-κB Activation
The upregulation of RAGE is followed by the activation of NF-κB pathway (Lander et al. 1997), thereby triggering a positive feedback loop in which RAGE expression is upregulated and thus the binding capacity of the AGEs is enhanced. As expected, activation of NF-κB pathway was characterized by selective induction of the nuclear translocation of p65 and p50 subunits (Valerio et al. 2006). We used western blot to determine the level of the p65 subunit in the nucleus. Interestingly, anti-RAGE antibody, PDTC and rosiglitazone treatment were able to block AGEs-induced NF-κB activation by decreasing p65 translocation from the cytosol to the nucleus (F(6,21) = 4.893, P < 0.05, Fig. 8a, b; (F(4,15) = 7.492, P < 0.05, Fig. 8c, d; F(5,18) = 8.252, P < 0.01, Fig. 8e, f).

Anti-RAGE antibody, PDTC and rosiglitazone attenuate AGEs-triggered NF-κB signaling. MBCECs were treated the same as those in Fig. 6. Nuclear extracts were prepared from MBCECs, nuclear NF-κB p65 and histone H3 as loading control were examined by Western blot using respective antibodies. Representative immunoblot of NF-κB p65 detected by Western blot (a, c, e), protein quantification was expressed as the ratio (in percentage) of Veh + Veh group (b, d, f). Values shown are expressed as mean ± SEM; n = 4. *P < 0.05, **P < 0.01 vs AGEs + Veh group
Anti-RAGE Antibody, PDTC and Rosiglitazone Reverse AGEs-Induced Downregulation of PPARγ
Previous reports have shown that activation of NF-κB may contribute to PPARγ downregulation. To investigate whether such downregulation of PPARγ is involved in protein complex formation of NF-κB with PPARγ, we performed coimmunoprecipitation assay with fresh protein extract of MBCECs. In this experiment, anti-p65 antibody effectively pulled down the PPARγ protein which confirmed the association of PPARγ with immunoprecipitating p65 and its presence in the immune complex (Fig. 9a). Then we hypothesized that NF-κB p65 and RAGE activation in MBCECs would induce a decrease in PPARγ level. Because the earlier parts of this study have showed that anti-RAGE antibody, PDTC or rosiglitazone treatment caused a significant decrease in both RAGE and nucleus NF-κB p65 levels in AGEs-treated MBCECs, we measured whether anti-RAGE antibody, PDTC and rosiglitazone restore the level of PPARγ in AGEs-treated MBCECs by Western blot. Consistent with our hypothesis, both PDTC and rosiglitazone reversed the reduced PPARγ levels in AGEs-induced MBCECs (F(4,15) = 5.871, P < 0.05, Fig. 9d, e; F(5,18) = 4.787, P < 0.05, Fig. 9f, g). However, anti-RAGE antibody also led to PPARγ increase in AGEs-treated MBCECs, but did not reach statistical significance (F(6,21) = 5.458, P > 0.05, Fig. 9a, b) compared with AGEs treatment group.

PDTC and rosiglitazone increase AGEs-downregulated PPARγ level. a MBCECs were subjected to immunoprecipitation using an anti-p65 antibody, and then subjected to SDS–PAGE and immunoblotted with an PPARγ antibody. Input (total protein extracts) was used as positive control and non-specific IgG antibody was used as negative control for immunoprecipitation. The blot data are representative of three separate experiments. Pretreatment of MBCECs with anti-RAGE antibody, PDTC or rosiglitazone for 2 h, and then MBCECs were incubated with AGEs. Nuclear extracts were prepared as Fig. 8. Nuclear PPARγ were determined in MBCECs by Western blot. Representative immunoblot of PPARγ detected by Western blot (b, d, f), protein quantification was expressed as the ratio (in percentage) of Veh + Veh group (c, e, g).Values shown are expressed as mean ± SEM; n = 4. *P < 0.05, **P < 0.01 vs AGEs + Veh group
Discussion
In the present study, we have shown that the RAGE-NF-κB signaling facilitates the entry of peripheral Aβ in the direction of blood-to-brain and downregulates the expression of PPARγ in an in vitro BBB model. We reversed such itinerary of Aβ through pretreatment of anti-RAGE, an antibody against RAGE, and PDTC, an NF-κB signaling inhibitor. We also used rosiglitazone, a commonly used PPARγ agonist as anti-diabetic drug, that also reversed the influx transport of Aβ across the BBB by upregulating the expression of PPARγ. Receptor for advanced glycation end products (RAGE) has been implicated in the pathology of AD including diabetic cognitive impairment and BBB dysfunction in numerous occasions. RAGE expression is found on microglial, and neuronal cells, as well as on the endothelial and smooth muscle cells in the CNS vasculature (Neeper et al. 1992; Schmidt et al. 2000; Yan et al. 1996). RAGE is also found in the astrocytes of AD animal model (Choi et al. 2014), and it contributes to cognitive dysfunction by means of astrocytic inflammation (Han et al. 2014). The level of RAGE is increased in both neurons and the endothelium in the context of AD (Yan et al. 1995), and is overexpressed in the hippocampus and frontal lobes of AD patients (Lue et al. 2001). As a mediator of Aβ transport (Jaeger et al. 2009; Yan et al. 1996), RAGE ferries Aβ from the periphery to the CNS across the BBB (Deane et al. 2003). The tight junctions of the BBB, which maintain the integrity of the BBB, are disrupted by Aβ (Strazielle et al. 2000) in concert with RAGE (Kook et al. 2012). RAGE-mediated breakdown of the tight junctions and trafficking of Aβ have also been shown in retinal pigment epithelia (Park et al. 2015).
The nuclear factor-kappa B (NF-κB) pathway, a pathway involved in inflammatory signaling, has been shown to exacerbate the incidence of AD (Lai et al. 2014; Tang et al. 2013). NF-κB is a multisubunit transcription factor that resides in the cytoplasm as an inactive complex, and, when activated, induces the expression of a number of genes including cell adhesion molecules, cell surface receptors, and cytokines. Expression of NF-κB p65 is found on the BBB (Pan et al. 2010; Stone et al. 2011). A signaling cascade jointly mediated by RAGE and NF-κB has been shown in diabetic vasculopathy. Overexpression of RAGE/NF-κB has also been shown in the hippocampus of diabetic animals (Xu et al. 2008). Aβ-mediated downregulation of p-glycoprotein, an efflux transporter at the BBB linked to AD pathology (Jeynes and Provias 2011), is also mediated by RAGE-NF-κB signaling (Park et al. 2015). Although the deleterious effect of RAGE-NF-κB signaling axis in the brain has been well documented, the signaling axis might also be involved in neuroprotection—it has been shown that the axis is crucial for neuronal differentiation of neural progenitor cells (NPCs), induced by several factors (Meneghini et al. 2013). Moreover, in a murine model of traumatic brain injury (TBI), cognitive recovery is correlated with the proneurogenic effects induced by intracerebroventricular administration of the RAGE ligand S100B (Kleindienst et al. 2005), possibly via the activation of RAGE-NF-κB axis (Bortolotto and Grilli 2016). These facts that both BBB breakdown and adult neurogenesis induction may rely on activation of RAGE-NF-κB signaling are likely to reflect high-level complexity within the pathways involved in these processes. PPARγ, a member of the nuclear receptor family, has been implicated in diabetes-related mitochondrial dysfunction (Yang et al. 2012). Incident of RAGE-mediated PPARγ downregulation has been documented in human chondrocytes (Ma et al. 2015; Chen et al. 2013). NF-κB signaling is a downregulator of PPARγ expression, and activation of PPARγ can block the activation of NF-κB (Nencioni et al. 2003) through the inhibition of NF-κB transcription factor (Kutsenko et al. 2012). TNFα, which is an inducer of NF-κB signaling, can degrade PPARγ through caspase-1 (He et al. 2008), which is regulated by RAGE (LeBlanc et al. 2014). Interrelating all these factors, we proposed that RAGE-NF-κB signaling may downregulate the expression of PPARγ and facilitate Aβ transport across the BBB into the brain. In our study, treatment with AGEs in MBCECs showed an increase in influx transport of Aβ. To find out the possible mechanisms, we blocked the activity of RAGE with its antibody that significantly inhibited the entry of Aβ from the luminal compartment to the abluminal compartment. The activity of RAGE was also suppressed by PDTC, an NF-κB inhibitor, suggesting a co-participation of RAGE and NF-κB in such environment. The same was further confirmed when pretreatment with anti-RAGE also affected the level of NF-κB p65 expression owing to a marked decrease in NF-κB p65 relative abundance. Next, to see whether the RAGE-NF-κB signaling loop had an inhibitory effect on PPARγ expression, we measured the expression of PPARγ in AGEs-treated MBCECs culture which was significantly decreased and such decrease in PPARγ expression was reversed by anti-RAGE and PDTC. Pretreatment with rosiglitazone, a PPARγ agonist, upregulated the expression of PPARγ and suppressed the inhibitory effect of RAGE and NF-κB, suggesting their role in downregulation of PPARγ. These findings collectively suggest that RAGE and NF-κB mediate influx transport of Aβ through downregulation of PPARγ, and that inhibition of RAGE-NF-κB signaling and promotion of PPARγ may have important implications for diabetes patients with cognitive impairment.
References
Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA (2004) Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol 61:661–666
Biessels GJ, Reagan LP (2015) Hippocampal insulin resistance and cognitive dysfunction. Nat Rev Neurosci 16:660–671
Bortolotto V, Grilli M (2016) Not only a bad guy: potential proneurogenic role of the RAGE/NF-κB axis in Alzheimer’s disease brain. Neural Regen Res 11:1924–1925
Chen TG, Chen TL, Chang HC, Tai YT, Cherng YG, Chang YT, Chen RM (2007) Oxidized low-density lipoprotein induces apoptotic insults to mouse cerebral endothelial cells via a Bax-mitochondria-caspase protease pathway. Toxicol Appl Pharmacol 219:42–53
Chen YJ, Sheu ML, Tsai KS, Yang RS, Liu SH (2013) Advanced glycation end products induce peroxisome proliferator-activated receptor γ down-regulation-related inflammatory signals in human chondrocytes via toll-like receptor-4 and receptor for advanced glycation end products. PLoS One 8:66611
Chen CJ, Ou YC, Li JR, Chang CY, Pan HC, Lai CY, Liao SL, Raung SL, Chang CJ (2014) Infection of pericytes in vitro by Japanese encephalitis virus disrupts the integrity of the endothelial barrier. J Virol 88:1150–1161
Chen F, Dong RR, Zhong KL, Ghosh A, Tang SS, Long Y, Hu M, Miao MX, Liao JM, Sun HB, Kong LY, Hong H (2016) Antidiabetic drugs restore abnormal transport of amyloid-β across the blood–brain barrier and memory impairment in db/db mice. Neuropharmacology 101:123–136
Choi BR, Cho WH, Kim J, Lee HJ, Chung C, Jeon WK, Han JS (2014) Increased expression of the receptor for advanced glycation end products in neurons and astrocytes in a triple transgenic mouse model of Alzheimer’s disease. Exp Mol Med 46:e75
Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B (2003) RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med 9:907–913
Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV (2004a) LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 43:333–344
Deane R, Wu Z, Zlokovic BV (2004b) RAGE (Yin) versus LRP1 (Yang) balance regulates Alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke 35:2628–2631
Fang Z, He QW, Li Q, Chen XL, Baral S, Jin HJ, Zhu YY, Li M, Xia YP, Mao L, Hu B (2016) MicroRNA-150 regulates blood–brain barrier permeability via Tie-2 after permanent middle cerebral artery occlusion in rats. FASEB J 30:2097–2107
Han C, Lu Y, Wei Y, Wu B, Liu Y, He R (2014) D-ribosylation induces cognitive impairment through RAGE-dependent astrocytic inflammation. Cell Death Dis 5:e1117
He F, Doucet JA, Stephens JM (2008) Caspase-mediated degradation of PPARgamma proteins in adipocytes. Obesity (Silver Spring) 16:1735–1741
Hong H, Lu Y, Ji ZN, Liu GQ (2006) Up-regulation of P-glycoprotein expression by glutathione depletion-induced oxidative stress in rat brain microvessel endothelial cells. J Neurochem 98:1465–1473
Hong H, Liu LP, Liao JM, Wang TS, Ye FY, Wu J, Wang YY, Wang Y, Li YQ, Long Y, Xia YZ (2009) Downregulation of LPR1 at the blood-brain barrier in streptozotocin-induced diabetic mice. Neuropharmacology 56:1054–1059
Hoyer S, Nitsch R (1989) Cerebral excess release of neurotransmitter amino acids subsequent to reduced cerebral glucose metabolism in early-onset dementia of Alzheimer type. J Neural Transm 75:227–232
Huber JD (2008) Diabetes, cognitive function, and the blood-brain barrier. Curr Pharm Des 14:1594–1600
Jaeger LB, Dohgu S, Sultana R, Lynch JL, Owen JB, Erickson MA, Shah GN, Price TO, Fleegal-Demotta MA, Butterfield DA, Banks WA (2009) Lipopolysaccharide alters the blood–brain barrier transport of amyloid β protein: a mechanism for inflammation in the progression of Alzheimer’s disease. Brain Behav Immun 203:507–517
Jeynes B, Provias J (2011) An investigation into the role of P-glycoprotein in Alzheimer's disease lesion pathogenesis. Neurosci Lett 487:389–393
Kleindienst A, McGinn MJ, Harvey HB, Colello RJ, Hamm RJ, Bullock MR (2005) Enhanced hippocampal neurogenesis by intraventricular S100B infusion is associated with improved cognitive recovery after traumatic brain injury. J Neurotrauma 22:645–655
Kook SY, Hong HS, Moon M, Ha CM, Chang S, Mook-Jung I (2012) Aβ1–42-RAGE interaction disrupts tight junctions of the blood–brain barrier via Ca2+-calcineurin signaling. J Neurosci 32:8845–8854
Kutsenko NL, Vesnina LE, Kaĭdashev IP (2012) Pioglitazone, an activator of PPAR-gamma, reduces the expression of kB nuclear factor and inhibits apoptosis in mononuclear cells of peripheral blood in vitro. Fiziol Zh 58:33–38
Lai J, Hu M, Wang H, Hu M, Long Y, Miao MX, Li JC, Wang XB, Kong LY, Hong H (2014) Montelukast targeting the cysteinyl leukotriene receptor 1 ameliorates Aβ1-42-induced memory impairment and neuroinflammatory and apoptotic responses in mice. Neuropharmacology 79:707–714
Lander HM, Tauras JM, Ogiste JS, Hori O, Moss RA, Schmidt AM (1997) Activation of the receptor for advanced glycation end products triggers a p21 (ras)-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J Biol Chem 272:17810–17814
LeBlanc PM, Doggett TA, Choi J, Hancock MA, Durocher Y, Frank F, Nagar B, Ferguson TA, Saleh M (2014) An immunogenic peptide in the A-box of HMGB1 protein reverses apoptosis-induced tolerance through RAGE receptor. J Biol Chem 289:7777–7786
Liu LP, Hong H, Liao JM, Wang TS, Wu J, Chen SS, Li YQ, Long Y, Xia YZ (2009) Upregulation of RAGE at the blood-brain barrier in streptozotocin-induced diabetic mice. Synapse 63:636–642
Liu Y, Liu F, Grundke-Iqbal I, Iqbal K, Gong CX (2011a) Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J Pathol:22554–22562
Liu Y, Ma Y, Wang R, Xia C, Zhang R, Lian K, Luan R, Sun L, Yang L, Lau WB, Wang H, Tao L (2011b) Advanced glycation end products accelerate ischemia/reperfusion injury through receptor of advanced end product/nitrativethioredoxin inactivation in cardiac microvascular endothelial cells. Antioxid Redox Signal 15:1769–1778
Liu LP, Yan TH, Jiang Y, Hu W, Hu M, Wang C, Zhang Q, Long Y, Wang JQ, Li YQ, Hu M, Hong H (2013) Pioglitazone ameliorates memory deficits in streptozotocin-induced diabetic mice by reducing brain β-amyloid through PPARγ activation. Acta Pharmacol Sin 34:455–463
Lue LF, Walker DG, Brachova L, Beach TG, Rogers J, Schmidt AM, Stern DM, Yan SD (2001) Involvement of microglial receptor for advanced glycation end products (RAGE) in Alzheimer’s disease: identification of a cellular activation mechanism. Exp Neurol 171:29–45
Ma C, Zhang Y, Li YQ, Chen C, Cai W, Zeng YL (2015) The role of PPARγ in advanced glycation end products-induced inflammatory response in human chondrocytes. PLoS One 10:e0125776
Meneghini V, Bortolotto V, Francese MT, Dellarole A, Carraro L, Terzieva S, Grilli M (2013) High-mobility group box-1 protein and β-amyloid oligomers promote neuronal differentiation of adult hippocampal neural progenitors via receptor for advanced glycation end products/nuclear factor-κB axis: relevance for Alzheimer’s disease. J Neurosci 33:6047–6059
Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, Elliston K, Stern D, Shaw A (1992) Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem 267:14998–15004. https://www.ncbi.nlm.nih.gov/pubmed/1378843
Nencioni A, Wesselborg S, Brossart P (2003) Role of peroxisome proliferator-activated receptor g and its ligands in the control of immune responses. Crit Rev Immunol 23:1–13
Nishitsuji K, Hosono T, Nakamura T, Bu G, Michikawa M (2011) Apolipoprotein E regulates the integrity of tight junctions in an isoform-dependent manner in an in vitro blood-brain barrier model. J Biol Chem 286:17536–17542
Ohtsuki S, Terasaki T (2007) Contribution of carrier-mediated transport systems to the blood–brain barrier as a supporting and protecting interface for the brain; importance for CNS drug discovery and development. Pharm Res 24:1745–1758
Pan W, Yu C, Hsuchou H, Kastin AJ (2010) The role of cerebral vascular NFκB in LPS-induced inflammation: differential regulation of efflux transporter and transporting cytokine receptors. Cell Physiol Biochem 25:623–630
Pardridge WM (2007) Blood-brain barrier delivery. Drug Discov Today 12:54–61
Park SW, Kim JH, Park SM, Moon M, Lee KH, Park KH, Park WJ, Kim JH (2015) RAGE mediated intracellular Aβ uptake contributes to the breakdown of tight junction in retinal pigment epithelium. Oncotarget 6:35263–35273
Pflanzner T, Janko MC, André-Dohmen B, Reuss S, Weggen S, Roebroek AJ, Kuhlmann CR, Park R, Kook SY, Park JC, Mook-Jung I (2014) Aβ1–42 reduces P-glycoprotein in the blood–brain barrier through RAGE–NF-κB signaling. Cell Death Dis 5:e1299
Provias J, Jeynes B (2014) The role of the blood-brain barrier in the pathogenesis of senile plaques in Alzheimer’s disease. Int J Alzheimers Dis 2014:191863
Sanz CM, Hanaire H, Vellas BJ, Sinclair AJ, Andrieu S, Study Group (2012) Diabetes mellitus as a modulator of functional impairment and decline in Alzheimer’s disease. The real. FR cohort. Diabet Med 29:541–548
Schmidt AM, Yan SD, Yan SF, Stern DM (2000) The biology of the receptor for advanced glycation end products and its ligands. Biochim Biophys Acta 1498:99–111
Schreck R, Meier B, Männel DN, Dröge W, Baeuerle PA (1992) Dithiocarbamates as potent inhibitors of nuclear factor κB activation in intact cells. J Exp Med 175:1181–1194
Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV (2000) Clearance of Alzheimer’s amyloid-ss (1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest 106:1489–1499
Shi L, Yu X, Yang H, Wu X (2013) Advanced glycation end products induce human corneal epithelial cells apoptosis through generation of reactive oxygen species and activation of JNK and p38 MAPK pathways. PLoS One 8:e66781
Stewart R, Liolitsa D (1999) Type 2 diabetes mellitus, cognitive impairment and dementia. Diabet Med 16:93–112
Stone KP, Kastin AJ, Pan W (2011) NFĸB is an unexpected major mediator of interleukin-15 signaling in cerebral endothelia. Cell Physiol Biochem 28:115–124
Strazielle N, Ghersi-Egea JF, Ghiso J, Dehouck MP, Frangione B, Patlak C, Fenstermacher J, Gorevic P (2000) In vitro evidence that β-amyloid peptide 1–40 diffuses across the blood–brain barrier and affects its permeability. J Neuropathol Exp Neurol 59:29–38
Tang SS, Wang XY, Hong H, Long Y, Li YQ, Xiang GQ, Jiang LY, Zhang HT, Liu LP, Miao MX, Hu M, Zhang TT, Hu W, Ji H, Ye FY (2013) Leukotriene D4 induces cognitive impairment through enhancement of CysLT1R-mediated amyloid-β generation in mice. Neuropharmacology 65:182–192. http://www.sciencedirect.com/science/article/pii/S002839081200473X
Ueno M, Nakagawa T, Wu B, Onodera M, Huang CL, Kusaka T, Araki N, Sakamoto H (2010) Transporters in the brain endothelial barrier. Curr Med Chem 17:1125–1138
Valerio A, Boroni F, Benarese M, Sarnico I, Ghisi V, Bresciani LG, Ferrario M, Borsani G, Spano P, Pizzi M (2006) NF-kappaB pathway: a target for preventing beta-amyloid (Abeta)-induced neuronal damage and Abeta42 production. Eur J Neurosci 23:1711–1720
Wan W, Cao L, Liu L, Zhang C, Kalionis B, Tai X, Li Y, Xia S (2015) Aβ1–42 oligomer-induced leakage in an in vitro blood–brain barrier model is associated with up-regulation of RAGE and metalloproteinases, and down-regulation of tight junction scaffold proteins. J Neurochem 134:382–393
Wuest DM, Lee KH (2012) Optimization of endothelial cell growth in a murine in vitro blood-brain barrier model. Biotechnol J 7:409–417
Xie Y, Ye L, Zhang X, Cui W, Lou J, Nagai T, Hou X (2005) Transport of nerve growth factor encapsulated into liposomes across the blood-brain barrier: in vitro and in vivo studies. J Control Release 105:106–119
Xu L, Li B, Cheng M, Zhang W, Pan J, Zhang C, Gao H (2008) Oral administration of grape seed proanthocyanidin extracts downregulate RAGE dependant nuclear factor-kappa BP65 expression in the hippocampus of streptozotocin induced diabetic rats. Exp Clin Endocrinol Diabetes 116:215–224
Yan SD, Yan SF, Chen X, Fu J, Chen M, Kuppusamy P, Smith MA, Perry G, Godman GC, Nawroth P et al (1995) Non-enzymatically glycated tau in Alzheimer’s disease induces neuronal oxidant stress resulting in cytokine gene expression and release of amyloid beta-peptide. Nat Med 1:693–699
Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM (1996) RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 382:685–691
Yang C, Aye CC, Li X, Diaz Ramos A, Zorzano A, Mora S (2012) Mitochondrial dysfunction in insulin resistance: differential contributions of chronic insulin and saturated fatty acid exposure in muscle cells. Biosci Rep 32:465–478
Yin KJ, Lee JM, Chen SD, Xu J, Hsu CY (2002) Amyloid-beta induces Smac release via AP-1/Bim activation in cerebral endothelial cells. J Neurosci 22:9764–9770. https://www.ncbi.nlm.nih.gov/pubmed/12427831
Youssef S, Nguyen DT, Soulis T, Panagiotopoulos S, Jerums G, Cooper ME (1999) Effect of diabetes and aminoguanidine therapy on renal advanced glycation end-product binding. Kidney Int 55:907–916
Zhang Y, Lee DH (2011) Sink hypothesis and therapeutic strategies for attenuating Abeta levels. Neuroscientist 17:163–173
Zlokovic BV (2008) The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57:178–201
Acknowledgements
We acknowledge the technical assistance of Dr. Feng Hao of XBL-China and Prof Yu Hui Xin of Jiangnan Institute of Nuclear Medicine. This work was supported by grants from the National Natural Science Foundation of China (81273497 to Hao Hong, 81603113 to Susu Tang), the Natural Science Foundation of Jiangsu Province (BK20150705 to Susu Tang), and the Program for Changjiang Scholars and Innovative Research Team in University (IRT1193), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Chen, F., Ghosh, A., Hu, M. et al. RAGE-NF-κB-PPARγ Signaling is Involved in AGEs-Induced Upregulation of Amyloid-β Influx Transport in an In Vitro BBB Model. Neurotox Res 33, 284–299 (2018). https://doi.org/10.1007/s12640-017-9784-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-017-9784-z