
Abstract
Streptozotocin has been widely used to mimic some aspects of Alzheimer’s disease (AD). However, especially in mice, several characteristics involved in the streptozotocin (STZ)-induced AD pathology are not well known. The main purpose of this study was to evaluate temporally the expression of AD-related proteins, such as amyloid-β (Aβ), choline acetyltransferase (ChAT), synapsin, axonal neurofilaments, and phosphorylated Tau in the hippocampus following intracerebroventricular (icv) administration of STZ in adult mice. We also analyzed the impact of STZ on short- and long-term memory by novel object recognition test. Male mice were injected with STZ or citrate buffer, and AD-related proteins were evaluated by immunoblotting assays in the hippocampus at 7, 14, or 21 days after injection. No differences between the groups were found at 7 days. The majority of AD markers evaluated were found altered at 14 days, i.e., the STZ group showed increased amyloid-β protein and neurofilament expression, increased phosphorylation of Tau protein, and decreased synapsin expression levels compared to controls. Except for synapsin, all of these neurochemical changes were transient and did not last up to 21 days of STZ injection. Moreover, both short-term and long-term memory deficits were demonstrated after STZ treatment at 14 and 21 days after STZ treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
First described by Alois Alzheimer in 1906, Alzheimer’s disease (AD) is characterized by the progressive loss of memory, cognitive impairment, decline of the language function, and several behavioral changes, including paranoia, delusions, and impaired social functioning. In advanced stages of the disease, patients might also present symptoms similar to those described in Parkinson disease, such as motor performance deficits and coordination difficulty (Selkoe 2001). Genetic and environmental factors seem to contribute to the development of AD (Kar et al. 2004). Age, traumatic brain injury, depression, exposure to toxic substances, and deficiency of neurotrophic factors may also be determinant for the development of this pathology (Muller-Spahn and Hock 1999).
The two main histopathological hallmarks of AD are the accumulation of extracellular insoluble fibrous material in the brain forming neuritic or senile plaques and the intracellular formation of neurofibrillary lesions (Braak and Braak 1996). The accumulation of both senile plaques and neurofibrillary lesions seem to exert an important role in the development of disease symptoms, possibly resulting in the degeneration of neurons with consequent reduction in the number of synapses (Goedert 1996).
Several abnormalities were also observed in the brain glucose metabolism of patients with sporadic AD. A primary disorder of neuronal insulin and a possibly impaired translation of insulin receptor may also contribute to the dementia development (Lannert and Hoyer 1998).
A large number of experimental models have been developed in the last two decades for investigation of AD-related mechanisms and evaluation of potential therapeutics. Most of these models are transgenic mouse models generated by overexpression of amyloid-β (Aβ) precursor protein and/or Tau (Chen et al. 2013). The transgenic mouse models have become promising tools to study the mechanisms involved in Tau phosphorylation and Aβ deposits; however, not all mice develop the extensive cell loss observed in human AD patients (Spires and Hyman 2005) and do not present all abnormalities seen in human AD (Chen et al. 2013; Spires and Hyman 2005). Moreover, there is a large discrepancy in the age of onset of pathology among those different AD models (it could appear from the age of 3 to 18 months, depending on the model) (Spires and Hyman 2005).
Many aspects of the AD were observed following the intracerebroventricular (icv) injections of streptozotocin (STZ). This compound is synthesized by Streptomycetes achromogenes, and it is commonly used in the systemic induction of diabetes due to its ability to damage the pancreatic β cells and to induce insulin resistance (Szkudelski 2001). STZ also inhibits the function of the insulin receptor in the brain, disrupting the metabolism of glucose and energy (Muller et al. 1998). Adult rats that received icv injections of STZ developed long-term progressive deficit of memory, learning, and cognitive behavior (Lannert et al. 1998). The icv injection of STZ induces accumulation of Aβ (Knezovic et al. 2015), increased Tau phosphorylation (Chen et al. 2013), and oxidative stress in animal brains, as revealed by increased levels of malondialdehyde and decreased levels of glutathione (Grunblatt et al. 2004; Tota et al. 2010). This model of sporadic AD is also characterized by progressive deterioration of cognitive function along with changes in metabolism of glucose and energy (Deng et al. 2009; Grunblatt et al. 2004; Lannert and Hoyer 1998; Lannert et al. 1998). However, STZ-induced AD model is not fully characterized, especially in mice, and requires more investigation.
The main purpose of this study was to evaluate temporally the expression of AD-related proteins, such as Aβ, choline acetyltransferase (ChAT), synaptic proteins, and axonal neurofilaments in the hippocampus following icv administration of STZ in adult mice. We also analyzed levels of Tau phosphorylation and the impact of STZ on hippocampus-dependent short- and long-term memory.
Materials and Methods
Animals
Ten-week-old male C57BL/6 mice (Jackson Laboratories, Maine, USA), weighing between 25 and 30 g, were used throughout this study. The animals had free access to food and water and were maintained on a 12:12 h light–dark cycle. All procedures were approved by the Institutional Animal Care Committee of the Institute of Biomedical Sciences, University of Sao Paulo, Brazil (protocol number: 098/2012).
Surgical Procedures
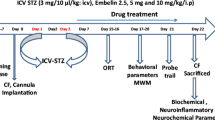
Mice were anesthetized using 5% isoflurane with oxygen as a carrier gas and maintained at 2–3% during surgery. The animals were placed into a stereotaxic frame with nose and ear bars especially designed for mice (Insight Ltda, SP, Brazil). STZ (3 mg/kg, icv) or citrate buffer (0.05 mol/L, pH 4.5) was injected bilaterally into the lateral ventricles. STZ (Sigma, St. Louis, MO) was dissolved in citrate buffer immediately before injection. The injection of STZ was performed using a Hamilton syringe (model 701) at the following coordinates: AP −0.5 mm; ML ±1.1 mm; DV −2.8 mm relative to the bregma. A total volume of 1.5 μl was injected in each hemisphere. The injection was conducted at a rate of 0.5 μL/min, and the needle was left in place for additional 3 min before it was slowly removed. The injection was repeated 2 days after the first STZ injection (1.5 mg/kg per day of injection). Additionally, control operated mice were infused bilaterally with 1.5 μl of vehicle (citrate buffer) into the lateral ventricles. Clinical signs were also monitored daily after the surgery, including general body condition and dehydration.
In order to confirm that the stereotaxic coordinates used throughout the study were suitable for STZ injection into lateral ventricles, FITC-labeled latex microspheres were injected into lateral ventricles and the presence of fluorescence in the surrounding brain tissue was examined 24 h after injection.
Immunoblotting
Animals were sacrificed by decapitation and the hippocampi were rapidly collected. To detect protein phosphorylation samples, n = 6–9 for each group were homogenized at 4 °C in extraction buffer (Tris, pH 7.4, 100 mM; sodium pyrophosphate 100 mM; sodium fluoride 100 mM; EDTA 10 mM, sodium orthovanadate 10 mM; PMSF 2 mM; aprotinin 0.01 mg/ml). For non-phosphorylated proteins (n = 5–8 for each group), the following buffer was used: Tris, pH 7.4, 100 mM; EDTA 10 mM; PMSF 2 mM; aprotinin 0.01 mg/ml. All the homogenates were centrifuged for 15 min at 12,000 rpm at 4 °C. The protein concentration of the supernatant was determined using the Bradford method (Bio-Rad, CA, USA). The samples were stored in sample buffer (Tris/HCl 500 mM, pH 6.8; 10% of SDS, 0.25% of bromophenol Blue; 10% of 2-mercaptoethanol and 50% glycerol) at −70 °C until use.
Thirty micrograms of protein was applied to 12% (amyloid β, Tau and Phosphorylated Tau) or 6.5% (ChAT, synapsin and PAN, anti-neurofilament antibody) acrylamide SDS gels (Bio-Rad, CA, USA). These proteins were then electrotransferred to nitrocellulose membranes (Millipore, Billerica, MA, USA) at 100 V for 80 min using a Trans-Blot cell. The membranes were then blocked for 2 h at room temperature with phosphate-buffered saline (PBS) containing 0.05% Tween-20 (TTBS) and 5% non-fat milk and incubated overnight at 4 °C with the following antibodies: monoclonal anti-rabbit amyloid β (1:1000; Santa Cruz Biotechnology, USA), monoclonal anti-rabbit synapsin (1:2000; Chemicon, USA), monoclonal anti-mouse neurofilaments (1:2000; Invitrogen, USA), monoclonal anti-mouse Tau (1:2000; Sigma Chemical Co., USA), monoclonal anti-rabbit phosphorylated Tau (Ser199/202) (1:2000; Sigma Chemical Co., USA), monoclonal anti-goat ChAT (1:500), and monoclonal anti-mouse β-actin (1:5000; Sigma Chemical Co., USA). The probed proteins were developed by using a chemiluminescent kit (ECL, Amersham Biosciences, NJ, EUA), and the bound antibodies were visualized using radiographic films. β-actin was used in all experiments as an internal control. The quantification of band intensity was performed with ImageJ (National Institutes of Health, USA).
Novel Object Recognition Test
Mice were tested (n = 10–14 for each group) in a novel object recognition test based on the innate tendency of rodents to differentially explore novel objects over familiar ones in an open field arena (Chen et al. 2014). The behavioral assay consisted of three phases: a habituation phase, a sample phase, and a test phase. In the habituation phase, mice were habituated to the open field arena (35 × 29 × 16 cm), where they were allowed to explore the apparatus for 10 min and then were taken back to their home cage. This procedure was repeated three times, with 30-min intervals. On the second day (sample phase), mice were allowed to explore two identical objects (A and B) arranged in a symmetric position from the center of the arena, for 10 min. The animals returned to their home cage for a retention period. After 1- and 24-h intertrial intervals (test phase), mice were tested in the same arena to explore two objects: a familiar object (A) and a novel object (C and D, respectively). One of the previously explored objects was placed in the same location as in the sample phase. The other familiar object was replaced by a novel object. Mice were allowed to explore both objects for 5 min. The total exploration time of the objects during the first 5 min was quantified in the sample and test phases (1 and 24 h). Data collection was performed using a video tracking system Pinnacle Studio (Pinnacle Systems, Mountain View, CA, USA). The arena and the objects were cleaned with 5% ethanol between tests. The following behaviors were considered as exploration of the objects: sniffing, licking, or touching the object with the nose or with the front legs or directing the nose to the object at a distance ≤1 cm.
The animal preference for the novel object was calculated as the ratio between the time spent to explore the new object (Tnew) over the total time spent exploring both object (Ttotal), known as discrimination index (DI = Tnew/Ttotal).
Statistical Analysis
Results are presented as the mean ± standard errors (SEMs). Statistical analyses of data were generated using GraphPad Prism, version 3.02 (GraphPad Software Inc., San Diego, CA, USA). Statistical comparison was performed using analysis of variance (ANOVA), followed by Dunnett’s test or Tukey’s test (only for Table 1). P ≤ 0.05 was considered statistically significant.
Results
Immunoblotting
Immunoblotting data revealed an increase of Aβ protein expression in the hippocampus after icv administration of STZ (F3, 14 = 4.25), as shown in Fig. 1a. The optical density increase was found significant at 14 days post-lesion (78%), and this effect appeared to decrease by day 21. Similar results were also observed regarding the analysis of PAN (F3, 14 = 5.01), an axonal neurofilament marker. Increased PAN optical density was also observed at day 14 (312%) post-STZ administration (Fig. 1b).

Effect of STZ on Aβ (a) and PAN (b) protein expression in the hippocampus. The graphs represent mean ratio of Aβ and PAN densitometric data in relation to β-actin. * P < 0.05 vs control (Dunnett’s test). STZ streptozotocin, Aβ β-amyloid, PAN axonal neurofilament marker
The protein levels of synapsin, a component of the pre-synaptic vesicle membrane, were found progressively decreased at days 14 (66%) and 21 (77.2%) after lesion (F3, 13 = 6.54) (Fig. 2a). ChAT protein levels were unaltered in STZ-treated mice (F3, 15 = 1.22) (Fig. 2b).

Effect of STZ on synapsin (a) and ChAT (b) protein levels in the hippocampus. The graphs represent mean ratio of synapsin and ChAT densitometric data in relation to β-actin. * P < 0.05 and *** p < 0.001 vs control (Dunnett’s test). STZ streptozotocin, ChAT choline acetyltransferase
No changes were observed in the protein levels of total Tau after STZ administration (F3, 17 = 0.04) (Fig. 3a); however, we found a marked increase in the levels of phosphorylated Tau (140%) in the hippocampus of STZ-treated mice 14 days post-lesion (F3, 14 = 3.75) (Fig. 3b, c).

Effect of STZ on total Tau protein expression and on Tau phosphorylation in the hippocampus. The graphs represent mean ratio of total tau and tau phosphorylation densitometric data in relation to β-actin. *P < 0.05 vs control (Dunnett’s test). STZ streptozotocin; pTau phosphorylated Tau
Novel Object Recognition Test
No differences were found between STZ-lesioned mice and controls in the total time of object exploration (both objects) during the different phases (Table 1).
We found that all mice (STZ-treated and controls) explored the two objects equally during the sample phase. STZ-treated mice spent less time exploring the novel object than the familiar object when compared to the control group following a 1-h delay, at 14 and 21 days post-lesion (F3, 30 = 16.79). No differences were found between STZ-treated mice and controls at 7 days post-lesion. The same pattern of results were obtained when memory retention was tested 24 h after the sample session (F3, 33 = 11.35).
Altogether, these results clearly indicated that STZ-treatment significantly impaired object recognition memory following 1- or 24-h delay, at 14 and 21 days post lesion (Fig. 4).

Effect of STZ on short-term and long-term memory in mice. Memory was assessed by novel object recognition test. Object discrimination during the test phase is presented as a discrimination index (time exploring the novel object/total time for exploring). ***P < 0.001 vs control (Dunnett’s test). STZ streptozotocin
Discussion
In the present study, we aimed at analyzing the effects of STZ on the expression of AD-related proteins and on hippocampus-dependent short- and long-term memory. One of the major histopathological hallmarks of AD is the presence of extracellular amyloid plaques, composed mainly by Aβ peptide. The extracellular deposition of this protein may induce neuronal death and is a major cause of AD (Li et al. 2007). Aβ levels were found elevated in the serum and in the brain of patients with AD (Price and Sisodia 1998). The results obtained in the present study regarding the Aβ protein expression in the hippocampus following STZ are in agreement with the literature. We found that the expression of Aβ was increased by 78% particularly 14 days after lesion. It has been suggested that the accumulation of Aβ plaques in the hippocampus might be related to the reduction of memory performance over time (Reitz et al. 2009). Since this brain region is essential for the formation and consolidation of new memories, it is assumed that the accumulation of Aβ contributes to the characteristic memory deficits in AD (Reitz et al. 2009).
AD also involves a substantial loss of elements of the cholinergic system. Despite the well-described changes in the cholinergic system induced by STZ, in the present study, we found that ChAT levels were not affected by STZ treatment in the hippocampus. Similarly, STZ did not induce any significant changes in hippocampal ChAT protein levels 30 days following the treatment in rats (Santos et al. 2012). One possible explanation is the fact that only 6% of the hippocampus nerve terminals are cholinergic (Perdahl et al. 1984), which suggests that alterations induced by this compound in the hippocampus may not be significant or easy to be determined by immunoblotting assays. Consistent with this hypothesis, hippocampal ChAT activity was found to be downregulated 3 weeks following icv STZ treatment in rats (Blokland and Jolles 1993), suggesting that activity assays might provide more reliable results in this particular case.
Synapsin is a phosphoprotein related to synaptic vesicles. Its state of phosphorylation is regulated by a variety of physiological and pharmacological manipulations that control the release of neurotransmitters. Decreased levels of synapsin were described in the hippocampus of patients with AD (Perdahl et al. 1984). Similarly, we found decreased synapsin levels 14 days after STZ injection and thereafter. Loss of synapses described in AD patients may be related to cognitive status found in these patients and may represent a structural reason for the decline of cognitive function (DeKosky and Scheff 1990).
Another common feature of AD is the presence of neurofibrillary tangles (Spires and Hyman 2005) composed primarily of abnormally modified Tau protein, neurofilaments, and other cytoskeletal proteins (Liu et al. 2011). Three neurofilament subunits have been described so far: NF-L, NF-M, and NF-H (Lee and Cleveland 1996). Abnormal distribution of NF-L was described in the hippocampus of patients with early AD (Nakamura et al. 1997). The protein levels of those three neurofilament subunits were also found elevated in brain and cerebrospinal fluid of patients with AD (Hu et al. 2002; Wang et al. 2001). The data obtained in the present study corroborate findings from previous studies. Neurofilament protein expression was significantly increased following STZ-induced lesions, when compared to the control group. In patients with AD, neurofilament proteins found in the neurofibrillary tangles, as well as Tau proteins have been described to be extensively phosphorylated, most likely due to an imbalance between the kinase and phosphatase activities (Liu et al. 2011). Increased expression and/or activity of some protein kinases such as glycogen synthase kinase 3β, dual-specific tyrosine (Y) regulated kinase 1A, P25/cyclin-dependent kinase 5, and mitogen-activated protein kinases has been observed in AD brains, in contrast with decreased activity of phosphatases such as PP1, PP2A, and PP5 (Chung 2009).
It has been previously demonstrated that STZ induces Tau phosphorylation at Ser 199/202 and Thr205, but not at Ser214, Thr231, Ser262, Ser262/356, Ser396/404, Ser 404, and Ser422, in the hippocampus of mice (Chen et al. 2013). In rats, Tau phosphorylation at Thr212, Ser396, and Ser199 have been demonstrated 21 days following STZ injections (Deng et al. 2009). In addition, Tau phosphorylation at Ser396 and Ser404 was found significantly increased in the hippocampus as a long-term consequence of the administration of icv STZ in rats (Grunblatt et al. 2007). We found that the levels of Tau phosphorylation at Ser199/202 were markedly increased at 14 days following icv STZ treatment, which is in agreement with previous published observations (Chen et al. 2013).
Several studies demonstrated that administration of STZ into the brain of wild-type mice and rats also causes learning and memory deficits (Liu et al. 2014; Muller et al. 2012; Pinton et al. 2010; Santos et al. 2015; Sharma et al. 2008; Shoham et al. 2007; Tota et al. 2010). We demonstrated here that STZ injection induced a delayed deficit in both short- and long-term object recognition memories, which was observed at 14 and 21 but not at 7 days post-lesion. No differences were found between STZ-lesioned mice and controls in the total time of object exploration during the sample phase, suggesting no deficit in locomotion or overall exploration levels in the lesioned mice. Memory deficits were observed in mice 3 (Santos et al. 2015) and 4 weeks (Muller et al. 2012) after icv STZ injection by object recognition test; and 1 (Pinton et al. 2010), 2 (Sharma et al. 2008) and 3 weeks (Tota et al. 2010) after icv STZ injection when mice were tested in the Morris water-maze task. In rats, memory function was intact at 2 weeks (Shoham et al. 2007), but not 3 (Liu et al. 2014) and 4 weeks (Shoham et al. 2007), after icv STZ injection in the object recognition test. Memory impairment was also observed 2 (Wang et al. 2016) and 3 weeks (Elcioglu et al. 2016; Majkutewicz et al. 2016) after icv STZ injection in the Morris water-maze task in rats. These data suggest distinct late onset in the STZ-induced memory impairment.
Conclusions
Our experiments revealed increased amyloid-β and neurofilament protein expression, increased phosphorylation of Tau, and decreased synapsin expression levels at 14 days after icv injection of STZ. Except for the synapsin levels, most of those alterations were transient. STZ-induced downregulation in synapsin levels was time-dependent. Moreover, short-term and long-term memory deficits were observed at both 14 and 21 days following STZ treatment. Altogether, these findings provide evidence that at 14 days after STZ treatment, neurodegenerative markers associated with AD are altered in mice.
References
Blokland A, Jolles J (1993) Spatial learning deficit and reduced hippocampal ChAT activity in rats after an ICV injection of streptozotocin. Pharmacol Biochem Behav 44:491–494
Braak H, Braak E (1996) Evolution of the neuropathology of Alzheimer’s disease. Acta Neurol Scand Suppl 165:3–12
Chen Y et al (2013) A non-transgenic mouse model (icv-STZ mouse) of Alzheimer’s disease: similarities to and differences from the transgenic model (3xTg-AD mouse). Mol Neurobiol 47:711–725. doi:10.1007/s12035-012-8375-5
Chen Y et al (2014) Intracerebroventricular streptozotocin exacerbates Alzheimer-like changes of 3xTg-AD mice1. Mol Neurobiol 49:547–562. doi:10.1007/s12035-013-8539-y
Chung SH (2009) Aberrant phosphorylation in the pathogenesis of Alzheimer’s disease. BMB Rep 42:467–474
DeKosky ST, Scheff SW (1990) Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol 27:457–464. doi:10.1002/ana.410270502
Deng Y, Li B, Liu Y, Iqbal K, Grundke-Iqbal I, Gong CX (2009) Dysregulation of insulin signaling, glucose transporters, O-GlcNAcylation, and phosphorylation of tau and neurofilaments in the brain: implication for Alzheimer’s disease. In: Am J Pathol, vol 175. vol 5. United States, pp 2089–2098. doi:10.2353/ajpath.2009.090157
Elcioglu HK, Aslan E, Ahmad S, Alan S, Salva E, Elcioglu OH, Kabasakal L (2016) Tocilizumab’s effect on cognitive deficits induced by intracerebroventricular administration of streptozotocin in Alzheimer’s model. Mol Cell Biochem 420:21–28. doi:10.1007/s11010-016-2762-6
Goedert M (1996) Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Ann N Y Acad Sci 777:121–131
Grunblatt E, Hoyer S, Riederer P (2004) Gene expression profile in streptozotocin rat model for sporadic Alzheimer’s disease. J Neural Transm 111:367–386. doi:10.1007/s00702-003-0030-x
Grunblatt E, Salkovic-Petrisic M, Osmanovic J, Riederer P, Hoyer S (2007) Brain insulin system dysfunction in streptozotocin intracerebroventricularly treated rats generates hyperphosphorylated tau protein. J Neurochem 101:757–770. doi:10.1111/j.1471-4159.2006.04368.x
Hu YY et al (2002) Elevated levels of phosphorylated neurofilament proteins in cerebrospinal fluid of Alzheimer disease patients. Neurosci Lett 320:156–160
Kar S, Slowikowski SP, Westaway D, Mount HT (2004) Interactions between beta-amyloid and central cholinergic neurons: implications for Alzheimer’s disease. J Psychiatry Neurosci 29:427–441
Knezovic A, Osmanovic-Barilar J, Curlin M, Hof PR, Simic G, Riederer P, Salkovic-Petrisic M (2015) Staging of cognitive deficits and neuropathological and ultrastructural changes in streptozotocin-induced rat model of Alzheimer’s disease. J Neural Transm (Vienna) 122:577–592. doi:10.1007/s00702-015-1394-4
Lannert H, Hoyer S (1998) Intracerebroventricular administration of streptozotocin causes long-term diminutions in learning and memory abilities and in cerebral energy metabolism in adult rats. Behav Neurosci 112:1199–1208
Lannert H, Wirtz P, Schuhmann V, Galmbacher R (1998) Effects of estradiol (−17beta) on learning, memory and cerebral energy metabolism in male rats after intracerebroventricular administration of streptozotocin. J Neural Transm 105:1045–1063
Lee MK, Cleveland DW (1996) Neuronal intermediate filaments. Annu Rev Neurosci 19:187–217. doi:10.1146/annurev.ne.19.030196.001155
Li M, Chen L, Lee DH, Yu LC, Zhang Y (2007) The role of intracellular amyloid beta in Alzheimer’s disease. In: Prog Neurobiol, vol 83. vol 3. England, pp 131–139. doi:10.1016/j.pneurobio.2007.08.002
Liu P, Zou LB, Wang LH, Jiao Q, Chi TY, Ji XF, Jin G (2014) Xanthoceraside attenuates tau hyperphosphorylation and cognitive deficits in intracerebroventricular-streptozotocin injected rats. Psychopharmacology 231:345–356. doi:10.1007/s00213-013-3240-4
Liu Q, Xie F, Alvarado-Diaz A, Smith MA, Moreira PI, Zhu X, Perry G (2011) Neurofilamentopathy in neurodegenerative diseases. Open Neurol J 5:58–62. doi:10.2174/1874205X01105010058
Majkutewicz I et al (2016) Dimethyl fumarate attenuates intracerebroventricular streptozotocin-induced spatial memory impairment and hippocampal neurodegeneration in rats. Behav Brain Res 308:24–37. doi:10.1016/j.bbr.2016.04.012
Muller AP et al (2012) Physical exercise exacerbates memory deficits induced by intracerebroventricular STZ but improves insulin regulation of H(2)O(2) production in mice synaptosomes. J Alzheimers Dis 30:889–898. doi:10.3233/jad-2012-112066
Muller D, Nitsch RM, Wurtman RJ, Hoyer S (1998) Streptozotocin increases free fatty acids and decreases phospholipids in rat brain. J Neural Transm 105:1271–1281
Muller-Spahn F, Hock C (1999) Risk factors and differential diagnosis of Alzheimer’s disease. Eur Arch Psychiatry Clin Neurosci 249(Suppl 3):37–42
Nakamura Y et al (1997) Abnormal distribution of neurofilament L in neurons with Alzheimer’s disease. Neurosci Lett 225:201–204
Perdahl E, Adolfsson R, Alafuzoff I, Albert KA, Nestler EJ, Greengard P, Winblad B (1984) Synapsin I (protein I) in different brain regions in senile dementia of Alzheimer type and in multi-infarct. Dementia. J Neural Transm 60:133–141
Pinton S, da Rocha JT, Zeni G, Nogueira CW (2010) Organoselenium improves memory decline in mice: involvement of acetylcholinesterase activity. In: Neurosci Lett, vol 472. vol 1. 2010 Elsevier Ireland Ltd, Ireland, pp 56–60. doi:10.1016/j.neulet.2010.01.057
Price DL, Sisodia SS (1998) Mutant genes in familial Alzheimer’s disease and transgenic models. Annu Rev Neurosci 21:479–505. doi:10.1146/annurev.neuro.21.1.479
Reitz C, Honig L, Vonsattel JP, Tang MX, Mayeux R (2009) Memory performance is related to amyloid and tau pathology in the hippocampus. J Neurol Neurosurg Psychiatry 80:715–721. doi:10.1136/jnnp.2008.154146
Santos DB et al (2015) Probucol mitigates streptozotocin-induced cognitive and biochemical changes in mice. Neuroscience 284:590–600. doi:10.1016/j.neuroscience.2014.10.019
Santos TO, Mazucanti CH, Xavier GF, Torrao AS (2012) Early and late neurodegeneration and memory disruption after intracerebroventricular streptozotocin. Physiol Behav 107:401–413. doi:10.1016/j.physbeh.2012.06.019
Selkoe DJ (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 81:741–766
Sharma B, Singh N, Singh M, Jaggi AS (2008) Exploitation of HIV protease inhibitor Indinavir as a memory restorative agent in experimental dementia. In: Pharmacol Biochem Behav, vol 89. vol 4. United States, pp 535–545. doi:10.1016/j.pbb.2008.02.012
Shoham S, Bejar C, Kovalev E, Schorer-Apelbaum D, Weinstock M (2007) Ladostigil prevents gliosis, oxidative-nitrative stress and memory deficits induced by intracerebroventricular injection of streptozotocin in rats. Neuropharmacology 52:836–843. doi:10.1016/j.neuropharm.2006.10.005
Spires TL, Hyman BT (2005) Transgenic models of Alzheimer’s disease: learning from animals. NeuroRx 2:423–437. doi:10.1602/neurorx.2.3.423
Szkudelski T (2001) The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol Res 50:537–546
Tota S, Awasthi H, Kamat PK, Nath C, Hanif K (2010) Protective effect of quercetin against intracerebral streptozotocin induced reduction in cerebral blood flow and impairment of memory in mice. Behav Brain Res 209:73–79. doi:10.1016/j.bbr.2010.01.017
Wang H, Cheng H, Che Z (2016) Ameliorating effect of luteolin on memory impairment in an Alzheimer’s disease model. Mol Med Rep 13:4215–4220. doi:10.3892/mmr.2016.5052
Wang J, Tung YC, Wang Y, Li XT, Iqbal K, Grundke-Iqbal I (2001) Hyperphosphorylation and accumulation of neurofilament proteins in Alzheimer disease brain and in okadaic acid-treated SY5Y cells. FEBS Lett 507:81–87
Acknowledgements
Work supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), University of Sao Paulo—Núcleo de Apoio à Pesquisa em Neurociência Aplicada (NAPNA) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). K.G. R, B.R.A., and M.S.H. were the recipients of fellowships from FAPESP. Thanks are due to Adilson S. Alves for the technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical Approval
All procedures performed in studies involving animals were in accordance with the ethical standards of the institution at which the studies were conducted. This article does not contain any studies with human participants performed by any of the authors.
Rights and permissions
About this article

Cite this article
Ravelli, K.G., Rosário, B.d.A., Camarini, R. et al. Intracerebroventricular Streptozotocin as a Model of Alzheimer’s Disease: Neurochemical and Behavioral Characterization in Mice. Neurotox Res 31, 327–333 (2017). https://doi.org/10.1007/s12640-016-9684-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-016-9684-7