
Abstract
Amyotrophic lateral sclerosis (ALS) is an idiopathic, fatal, neurodegenerative disease of the human motor system. The pathogenesis of ALS is a topic of fascinating speculation and experimentation, with theories revolving around intracellular protein inclusions, mitochondrial structural issues, glutamate excitotoxicity and free radical formation. This review explores the rationale for the involvement of a novel protein, B-cell lymphoma/leukaemia 11b (Bcl11b) in ALS. Bcl11b is a multifunctional zinc finger protein transcription factor. It functions as both a transactivator and genetic suppressor, acting both directly, binding to promoter regions, and indirectly, binding to promoter-bound transcription factors. It has essential roles in the differentiation and growth of various cells in the central nervous system, immune system, integumentary system and cardiovascular system, to the extent that Bcl11b knockout mice are incompatible with extra-uterine life. It also has various roles in pathology including the suppression of latent retroviruses, thymic tumourigenesis and neurodegeneration. In particular its functions in neurodevelopment, viral latency and T-cell development suggest potential roles in ALS pathology.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis is a catastrophic disease for individuals, their families, friends and carers. The current lack of understanding of its pathogenesis engenders great difficulties in its diagnosis and treatment. ALS is a progressive, fatal illness that encompasses destruction of all components of the motor system along the neuraxis. While motor-system pathology is the most salient in the clinical presentation, co-occurrence of other neurodegenerative diseases such as frontotemporal dementia is seen in a number of cases (Li et al. 2012). At present, no definitive biomarker for ALS exists with clinicians relying upon clinical signs alone, using the El Escorial criteria to direct diagnosis (Kiernan et al. 2011). The incidence worldwide is around 2 per 100,000 population per year, and the population prevalence is approximately 6 in 100,000, with a slight male preponderance. From the onset of symptoms, diagnosis takes an average of 16 months (Donaghy et al. 2008) and the average survival from diagnosis is 2–3 years (Mitchell and Borasio 2007). Many theories on the aetiology of ALS have been put forward and several transgenic mouse models of ALS have established causative genetic links, including mutations of SOD1 gene, increased CCGGGG hexanucleotide repeats and most recently, up regulation of human endogenous retrovirus-K (HERV-K) (Wenxue Li et al. 2015). However, the common disease pathways of these disparate abnormalities is unknown (Kiernan et al. 2011; Mitchell and Borasio 2007). Improving patient outcomes in ALS fundamentally depends on developing an understanding of the disease mechanisms, which will catalyse development of early, specific diagnostic and therapeutic measures. Apart from supportive care, the only registered therapy is riluzole which improves life expectancy by only 2–3 months and has limited ability to abate functional decline (Zoccolella et al. 2007).
Bcl11b: Structure and Function
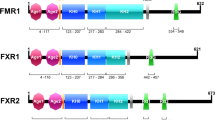
The functions of Bcl11b and the pathogenesis of amyotrophic lateral sclerosis have several connections. B-cell lymphoma/leukaemia 11b (BCL11b), also known as Coup-TF interacting protein 2 (CTIP2) and radiation-induced tumour suppressor gene 1 (RIT1), is a Kruppel-like C2H2 zinc finger protein transcription factor located on chromosome 14 in humans. The protein was first discovered as a tumour suppressor gene-related closely to Bcl11a (Satterwhite et al. 2001). Bcl11b has two splice isoforms, alpha and beta, (Fig. 1) comprising 823 and 894 amino acids, respectively, in humans. Exon 4 contains the six C2H2 zinc finger-binding domains with domains 3 and 4 binding to the DNA. It also contains regions for protein interaction (Fig. 2). Murine Bcl11b is located on chromosome 12 and has 88 % identity to that of humans (Huang et al. 2012). Bcl11b operates directly by binding to specific promoter regions or indirectly by binding to promoter-bound transcription factors Cismasiu et al. (2005). In its role as a transcription factor Bcl11b acts as both a repressor and transactivator of a myriad of genes. Bcl11b is an essential developmental transcription factor in a multiplicity of systems and has known roles in the various pathologies outlined in this review.

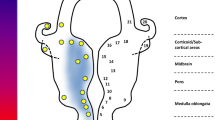
Schematic diagram showing the multifactorial, interrelated molecular and genetic pathways underlying neurodegeneration in ALS adapted from Kiernan et al. 2011

Schematic diagram showing the genetic structure of the two isoforms as well as the protein structure of the alpha isoform (with locations of 6 zinc finger proteins) adapted from Huang et al. 2012
Amyotrophic Lateral Sclerosis and Bcl11b
There are a number of key Bcl11b functions that are of relevance to ALS: CNS development, because of the neurodegenerative component to ALS; retroviral suppression, because of causative links between HERV-K and ALS; and T-cell development, because of inflammatory involvement in the disease. This review draws together the extant connections between the pathogenesis of ALS and the functions of Bcl11b, elucidating directions for future research.
Neuronal Degeneration
Bcl11b is expressed in the neocortex, hippocampus, striatum and vomeronasal system of humans and mice serving a number of functions. In the neocortex, it has been shown to play a role in directing axonal growth and path finding of corticospinal motor neurons (CSMNs). CSMNs are pyramidal, first-order motor neurons that connect to spinal motor neurons to give voluntary control of the skeletal muscles (Chen et al. 2004). Bcl11b is highly expressed in developing CSMNs of cortical layer V while at the same layer is absent from cortico-cortical neurons (Fame et al. 2012). Chen et al. found that Bcl11b acts downstream of Fezf2 to determine whether cortical neurons will project cortically or subcortically (B. Chen et al. 2008). Bcl11b deficient mice perish on day 1 post-natally, displaying severe abnormalities in fasciculation, outgrowth and path finding of axonal projections of CSMNs. To a lesser extent heterozygote, Bcl11b ± mice demonstrate reduced ability to correctly regulate and prune axons (Arlotta et al. 2005). During embryo development, the challenge for CSMNs is to extend axons over great lengths to very specific positions within the spinal cord. Bcl11b has been indirectly associated with a number of communicating, transmembrane proteins that are involved in this pathfinding including Contactin 6, Cadherin 13 and Cadherin 22, (Abbas et al. 2014). Bcl11b is a known regulator of the Notch signalling pathway which determines the expression of the aforementioned proteins (Betancourt et al. 2014). Bcl11b is also expressed in the granule cells of the hippocampus (Simon et al. 2012), medium spiny neurons of the striatum (Desplats et al. 2008) and sensory neurons of the olfactory bulb (Enomoto et al., 2011), playing roles in cellular differentiation and structural organisation in these cells. The core pathogenetic mechanism of ALS is the apoptosis of CSMNs (Cluskey and Ramsden 2001). As discussed previously, Bcl11b is a specific marker for CSMNs in the motor cortex and thus in ALS pathology Bcl11b may be elevated in CSF and serum as a consequence of apoptosis of CSMNs (Arlotta et al. 2005; Canovas et al. 2015).
Alternatively, similar to the neurodegeneration seen in Huntington’s Disease (HD), Bcl11b deficiency, resulting in loss of function, may be part of a causal disease pathway (Desplats et al. 2008). Bc11b has been found to be significantly decreased in HD-cell models, mouse models and human samples. The toxic effects of huntingtin protein are attenuated when Bcl11b is overexpressed (Desplats et al. 2008). Bcl11b binds to the proximal promoter region of multiple striatally enriched genes such as Klf9, Dgke, Pde10a and Isl1. Desplats et al. suggested that the decrease of the striatal gene expression caused by Bcl11b insufficiency may underlie Huntington’s pathology. Bcl11b has similar functions in promoting expression of CSMN specific proteins and directing axonal growth. A loss of Bcl11b function may contribute to motor neuron degeneration in a similar fashion to the striatal neuron degeneration seen in Huntington’s Disease.
Neuronal Regeneration
Bcl11b is an important regulator of neuronal cell growth. It may be up-regulated as part of an attempted regenerative process in ALS patients. ChIP-Seq transcriptome analysis of striatal cell lines reveals that Bcl11b regulates a myriad of brain-derived neurotrophic factor (BDNF) signalling pathway genes (Tang et al. 2011). BDNF is a member of the neurotrophin family which consists of nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin 3 (NT-3) and neurotrophin 4 (NT-4) (Zuccato and Cattaneo 2009). These trophic factors are essential to the development, differentiation and survival of CNS cells. Accordingly, altered activities of BDNF and other neurotrophins have been implicated in several neurodegenerative diseases, including Huntington’s, Alzheimer’s and Parkinson’s diseases. In the ChIP-Seq analysis, the majority of expression changes were decreases, suggesting that Bcl11b and BDNF function antagonistically; BDNF promotes synaptogenesis and axonal progression while Bcl11b negatively regulates synaptogenesis and directs axonal progression to ensure well-structured development. The question is then whether or not reparative neurogenesis occurs in ALS. Histopathological studies reveal that injury is repaired by astrogliosis (Cluskey and Ramsden 2001). In the mammalian adult brain, neurogenesis is normally restricted to two evolutionarily primitive regions: the olfactory bulb and the subventricular zone (SVZ) (Doetsch et al. 1997), and the hippocampal dentate gyrus (Pan et al. 2013). Fascinatingly Chen et al. demonstrated that following synchronous apoptosis of CSMNs, endogenous precursors migrated and differentiated into adult neurons. These new CSMNs redeveloped connections to the spinal cord from the cortical regions, forming such long-distance projections over a >12-week period (J. Chen et al. 2004). There has long been speculation and experimentation on the possibility of exogenous stem cell implantation to improve reparative neurogenesis (Mitchell and Borasio 2007) and thus if Bcl11b does play a role in the repair of motor neurons post injury it will be important part of our understanding of the molecular specifics of motor neuron regeneration.
Suppression of Endogenous Retroviruses
A large fraction of the human genome is comprised latent retroviruses integrated into the germline over several million years of evolution with some studies suggesting that these human endogenous retroviruses (HERVs) make up around 7 % of the genome (Bock and Stoye 2000; Löwer 1999). While there is no conclusive evidence for the pathogenicity of HERVs, a number of hypotheses have been proffered (Bock and Stoye 2000; Kolson and Gonzalez-Scarano 2001). HERVs may pathologically up regulate the transcription of gene sequences downstream to the HERV long-terminal repeats (LTRs), which contain the promoter elements of the viruses. HERVs may also produce super antigens that induce magnified host inflammatory responses. Alternatively exogenous viruses could initiate immune responses against comparable host HERVs leading to autoimmune phenomena seen in SLE or MS (Bock and Stoye 2000).
The function of Bcl11b in suppressing HERVs is an area for further investigation particularly given the presence of HERVs in diseases such as MS, ALS and systemic lupus erythematosis. HERVs are highly expressed in ALS tissue samples (Alfahad and Nath 2013), and Bcl11b may play a role in the suppression of such retroviruses similar to its role as a transcriptional silencer in HIV. Desplats et al. found that Bcl11b was significantly elevated in the CSF of latent HIV patients and was highly correlated with levels of HP1-alpha, MeCP2 and HDAC1, which are histone modifiers that act as HIV silencers. Immunostaining of human HIV-patient post-mortem tissue found Bcl11b was present in astrocytes and microglia (Desplats et al. 2013).
Bcl11b has a dual action in suppressing the activation of latent HIV-1. Acting co-operatively with the NuRD co-repressor complex, Bcl11b transcriptionally silences the HIV-1 long-terminal repeat, thus preventing the synthesis of the HIV TAT protein which is the key enhancer of viral transcription (Marban et al. 2005). Bcl11b also limits the reactivation of HIV-1 in the absence of TAT, indicating that it acts on endogenous transcription factors such as positive transcription elongation factor β (P-TEFβ) (Cherrier et al. 2013). The Bcl11b-NuRD complex induces transcriptional silencing through histone deacetylation and heterochromatin formation in the LTR HIV1 sequence (Cismasiu et al. 2008). It is this heterochromatin formation at the promoter region that suppresses retroviral activation in HIV and is thought to be involved in latency of other viruses (Le Douce et al. 2014).
Both HIV1 and HTLV1 can cause an ALS-like syndrome that, in the case of HIV1, radically improves with antiretroviral therapy, confirming that retroviruses have the capacity to cause motor neuron pathology (Moulignier et al. 2001; Verma and Berger 2006). Strikingly, even for some ALS patients without exogenous retroviral involvement there are radical increases in reverse transcriptase activity (RT), with early studies showing RT was present in the serum of 59 % of ALS patients versus 5 % of controls (Andrews et al. 2000). This was corroborated by MacGowan et al. who found RT involvement in the serum of 53 % of sporadic ALS patients compared with 12.25 % in non-related healthy controls (Alfahad and Nath 2013; MacGowan et al. 2007).
Given that the vast majority of the positive patients had not been infected with exogenous retroviruses, the purported source of the RT are HERVs. Indeed, analysis of serum samples and post-mortem brain tissue from ALS patients revealed significantly increased expression of HERV-K, compared to controls (Alfahad and Nath 2013). Importantly, HERV-K immunostaining was present in clusters of neurons but not in other cell types in the brain (Wanhe Li et al. 2012). Excitingly, a recent study by Li et al. found that in vitro cultures of human neurons that up-regulated HERV-K, either whole virus or env protein, induced pathological changes including retraction and beading of neurites. Further, transgenic mice highly expressing the HERV-K env gene developed an ALS phenotype characterized clinically by progressive motor dysfunction and histologically by selective decreases of motor cortex volume, diminished synaptic activity in pyramidal neurons, dendritic spine abnormalities, nucleolar dysfunction and DNA damage. Expression of HERV-K was negatively regulated by TARDBP43, which binds to the long-terminal repeat region of the virus (Wenxue Li et al. 2015). TARDBP43, similar to Bcl11b, is a transcription factor and suppresses HIV1 in human microglia by binding to the LTR and blocking the assembly of transcription complexes that function with TAT (Kato et al. 1991; Ou et al. 1995). This works in parallel to Bcl11b, which also acts on the LTR and induces heterochromatic formation.
Both Bcl11b and TARDBP43 function as responsive viral suppressors and have been well characterized in the context of HIV1. Given that TARDBP43 is known to also suppress HERV-K in ALS, there is a strong suggestion that Bcl11b may serve a similar adaptive function in ALS by suppressing transcription of pathogenic HERVs.
T-Cell Development and Neuroinflammation
Bcl11b may also be elevated as part of an immune or inflammatory component to ALS. In the immune system, Bcl11b is specifically and restrictively expressed in T cells. It is integral for the maturation and specification of αβ T cells. Bcl11b -/- precursor T cells show developmental arrest at the immature double-negative 2 stage (CD4-, CD8-) (Kominami 2012; Liu et al. 2010). While neuroinflammation in ALS is primarily driven by microglia and astrocytes (Lewis et al. 2012), infiltrating T-cell subpopulations modulate inflammatory process throughout the disease course (Philips and Robberecht 2011). T cells in the CNS are seldom found in early disease, but increase in frequency as the disease progresses (Chiu et al. 2008). Investigations into the role of T cells in neuroinflammation in the mSOD mouse suggest that these cells influence the phenotypic profile of activated microglia, with CD4+ T cells promoting an M2-type neuroprotective phenotype in microglia (Beers et al. 2008). When T cells are depleted in SOD1-mutant mice, the disease course (but not onset) is significantly worsened indicating that the inflammatory process surprisingly may in fact be neuroprotective (Henkel et al. 2009). Bcl11b has a role in the differentiation and maintenance of T-cell identity, and thus it may be elevated in the CNS and serum of ALS patients as part of the T-cell infiltrate and neuroprotective inflammatory process.
Directions for Future Research
It has been well established that Bcl11b is a specific marker for corticospinal projection motor neurons in layer 5 of the cerebral cortex (Arlotta et al. 2005; Canovas et al. 2015; Lodato et al. 2014). The recent Li et al. study found in immunohistochemistry of post-mortem mouse brains that there was a significant decrease of Bcl11b expressing cells in the motor cortex of transgenic, ALS phenotype mice when compared with healthy controls. By comparing, NeuN (a mature neuronal biomarker) in the frontal cortex and Satb2 (a marker of callosal projection neurons) in the motor cortex showed no significant changes in the number of immunopositive cells (Wenxue Li et al. 2015). This has clearly indicated that Bcl11b expressing cells are specifically affected in ALS pathology but it is unclear as to whether it has a functional involvement or is merely incidental to the disease.
Further to this, there has been only a small genetic link found in systematic mutation screening which revealed 1 ALS patient with a Bcl11b mutation of C.36A > T, coding Glu32Val, in a cohort of 190 ALS patients. The mutation was predicted to have a loss of function effect and potentially contribute to the disease progression (Daoud et al. 2011).
Conclusion
Bcl11b is a complex multifunctional protein with important roles in the CNS, immune, integumentary and other systems. Bcl11b deficiency in an organism causes death on day 1 post-natally and in various regions it induces pathologies of particular systems. Bcl11b has a number of potential roles to play in ALS and is an important target for future research.
References
Abbas S, Sanders MA, Zeilemaker A, Geertsma-Kleinekoort WMC, Koenders JE, Kavelaars FG, Abbas ZG, Mahamoud S, Chu IW, Hoogenboezem R, Peeters JK, van Drunen E, van Galen J, Beverloo HB, Löwenberg B, Valk PJM (2014) Integrated genome-wide genotyping and gene expression profiling reveals BCL11B as a putative oncogene in acute myeloid leukemia with 14q32 aberrations. Haematologica 99(5):848–857. doi:10.3324/haematol.2013.095604
Alfahad T, Nath A (2013) Retroviruses and amyotrophic lateral sclerosis. Antiviral Res 99(2):180–187. doi:10.1016/j.antiviral.2013.05.006
Andrews WD, Tuke PW, Al-Chalabi A, Gaudin P, Ijaz S, Parton MJ, Garson JA (2000) Detection of reverse transcriptase activity in the serum of patients with motor neurone disease. J Med Virol 61(4):527–532. doi:10.1002/1096-9071(200008)61:4<527:AID-JMV17>3.0.CO;2-A
Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, MacKlis JD (2005) Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron 45:207–221. doi:10.1016/j.neuron.2004.12.036
Beers DR, Henkel JS, Zhao W, Wang J, Appel SH (2008) CD4 + T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc Natl Acad Sci USA 105(40):15558–15563. doi:10.1073/pnas.0807419105
Betancourt J, Katzman S, Chen B (2014) Nuclear factor one B regulates neural stem cell differentiation and axonal projection of corticofugal neurons. J Comp Neurol 522:6–35. doi:10.1002/cne.23373
Bock M, Stoye JP (2000) Endogenous retroviruses and the human germline. Curr Opin Genet Dev 10(6):651–655. doi:10.1016/S0959-437X(00)00138-6
Canovas J, Berndt FA, Sepulveda H, Aguilar R, Veloso FA, Montecino M, Oliva C, Maass JC, Sierralta J, Kukuljan M (2015) The specification of cortical subcerebral projection neurons depends on the direct repression of TBR1 by CTIP1/BCL11a. J Neurosci 35(19):7552–7564. doi:10.1523/JNEUROSCI.0169-15.2015
Chen J, Magavi SSP, Macklis JD (2004) Neurogenesis of corticospinal motor neurons extending spinal projections in adult mice. Proc Natl Acad Sci USA 101(46):16357–16362. doi:10.1073/pnas.0406795101
Chen B, Wang SS, Hattox AM, Rayburn H, Nelson SB, McConnell SK (2008) The Fezf2-Ctip2 genetic pathway regulates the fate choice of subcortical projection neurons in the developing cerebral cortex. Proc Natl Acad Sci USA 105(32):11382–11387. doi:10.1073/pnas.0804918105
Cherrier T, Le Douce V, Eilebrecht S, Riclet R, Marban C, Dequiedt F, Goumon Y, Paillart JC, Mericskay M, Parlakian A, Bausero P, Abbas W, Herbein G, Kurdistani SK, Grana X, Van Driessche B, Schwartz C, Candolfi E, Benecke AG, Van Lint C, Rohr O (2013) CTIP2 is a negative regulator of P-TEFb. Proc Natl Acad Sci USA 110(31):12655–12660. doi:10.1073/pnas.1220136110
Chiu IM, Chen A, Zheng Y, Kosaras B, Tsiftsoglou SA, Vartanian TK, Brown RH Jr, Carroll MC (2008) T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc Natl Acad Sci USA 105(46):17913–17918. doi:10.1073/pnas.0804610105
Cismasiu VB, Adamo K, Gecewicz J, Duque J, Lin Q, Avram D (2005) BCL11B functionally associates with the NuRD complex in T lymphocytes to repress targeted promoter. Oncogene 24:6753–6764. doi:10.1038/sj.onc.1208904
Cismasiu VB, Paskaleva E, Suman Daya S, Canki M, Duus K, Avram D (2008) BCL11B is a general transcriptional repressor of the HIV-1 long terminal repeat in T lymphocytes through recruitment of the NuRD complex. Virology 380:173–181. doi:10.1016/j.virol.2008.07.035
Cluskey S, Ramsden DB (2001) Mechanisms of neurodegeneration in amyotrophic lateral sclerosis. Mol Pathol 54(6):386–392. doi:10.1136/mp.54.6.386
Daoud H, Valdmanis PN, Gros-Louis F, Belzil V, Spiegelman D, Henrion E, Diallo O, Desjarlais A, Gauthier J, Camu W, Dion PA, Rouleau GA (2011) Resequencing of 29 candidate genes in patients with familial and sporadic amyotrophic lateral sclerosis. Arch Neurol 68(5):587–593. doi:10.1001/archneurol.2010.351
Desplats PA, Lambert JR, Thomas EA (2008) Functional roles for the striatal-enriched transcription factor, Bcl11b, in the control of striatal gene expression and transcriptional dysregulation in Huntington’s disease. Neurobiol Dis 31:298–308. doi:10.1016/j.nbd.2008.05.005
Desplats P, Dumaop W, Smith D, Adame A, Everall I, Letendre S, Ellis R, Cherner M, Grant I, Masliah E (2013) Molecular and pathologic insights from latent HIV-1 infection in the human brain. Neurology 80(15):1415–1423. doi:10.1212/WNL.0b013e31828c2e9e
Doetsch F, García-Verdugo JM, Alvarez-Buylla A (1997) Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. J Neurosci 17(13):5046–5061
Donaghy C, Dick A, Hardiman O, Patterson V (2008) Timeliness of diagnosis in motor neurone disease: a population-based study. Ulster Med J 77(1):18–21
Enomoto T, Ohmoto M, Iwata T, Uno A, Saitou M, Yamaguchi T, Kominami R, Matsumoto I, Hirota J (2011) Bcl11b/Ctip2 controls the differentiation of vomeronasal sensory neurons in mice. J Neurosci 31(28):10159–10173. doi:10.1523/JNEUROSCI.1245-11.2011
Fame R, MacDonald J, Macklis J (2012) Development, specification, and diversity of callosal projection neurons. Trends Neurosci 29(6):997–1003. doi:10.1016/j.biotechadv.2011.08.021.Secreted
Henkel JS, Beers DR, Zhao W, Appel sh (2009) Microglia in ALS: the good, the bad, and the resting. J Neuroimmune Pharmacol 4(4):389–398. doi:10.1007/s11481-009-9171-5
Huang X, Du X, Li Y (2012) The role of BCL11B in hematological malignancy. Exp Hematol Oncol 1(1):22. doi:10.1186/2162-3619-1-22
Kato H, Horikoshi M, Roeder RG (1991) Repression of HIV-1 transcription by a cellular protein. Science 251(5000):1476–1479
Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC (2011) Amyotrophic lateral sclerosis. Lancet 377(9769):942–955. doi:10.1016/S0140-6736(10)61156-7
Kolson D, Gonzalez-Scarano F (2001) Endogenous retroviruses and multiple sclerosis. Ann Neurol 50(4):429–430
Kominami R (2012) Role of the transcription factor Bcl11b in development and lymphomagenesis. Proc Jpn Acad Ser B 88(3):72–87. doi:10.2183/pjab.88.72
Le Douce V, Cherrier T, Riclet R, Rohr O, Schwartz C (2014) CTIP2, a multifunctional protein: cellular physiopathology and therapeutic implications. Med Sci 30(8–9):797–802. doi:10.1051/medsci/20143008019
Lewis C-A, Manning J, Rossi F, Krieger C (2012) The neuroinflammatory response in ALS: the roles of microglia and T cells. Neurol Res Int 2012:1–8. doi:10.1155/2012/803701
Li W, Jin Y, Prazak L, Hammell M, Dubnau J (2012) Transposable elements in TDP-43-mediated neurodegenerative disorders. PLoS One 7(9):1–10. doi:10.1371/journal.pone.0044099
Li W, Lee M, Henderson L, Tyagi R, Bachani M, Steiner J, Campanac E, Hoffman DA, von Geldern G, Johnson K, Maric D, Morris HD, Lentz M, Pak K, Mammen A, Ostrow L, Rothstein J, Nath A (2015) Human endogenous retrovirus-K contributes to motor neuron disease. Science Translational Medicine 7(307):307ra153. doi:10.1126/scitranslmed.aac8201
Liu P, Li P, Burke S (2010) Critical roles of Bcl11b in T-cell development and maintenance of T-cell identity. Immunol Rev 238(1):138–149. doi:10.1111/j.1600-065X.2010.00953.x
Lodato S, Molyneaux BJ, Zuccaro E, Goff LA, Chen H-H, Yuan W, Meleski A, Takahashi E, Mahony S, Rinn JL, Gifford DK, Arlotta P (2014) Gene co-regulation by Fezf2 selects neurotransmitter identity and connectivity of corticospinal neurons. Nat Neurosci 17(8):1046–1054. doi:10.1038/nn.3757
Löwer R (1999) The pathogenic potential of endogenous retroviruses: facts and fantasies. Trends Microbiol 7(9):350–356. doi:10.1016/S0966-842X(99)01565-6
MacGowan DJL, Scelsa SN, Imperato TE, Liu K-N, Baron P, Polsky B (2007) A controlled study of reverse transcriptase in serum and CSF of HIV-negative patients with ALS. Neurology 68(22):1944–1946. doi:10.1212/01.wnl.0000263188.77797.99
Marban C, Redel L, Suzanne S, Van Lint C, Lecestre D, Chasserot-Golaz S, Leid M, Aunis D, Schaeffer E, Rohr O (2005) COUP-TF interacting protein 2 represses the initial phase of HIV-1 gene transcription in human microglial cells. Nucleic Acids Res 33(7):2318–2331. doi:10.1093/nar/gki529
Mitchell JD, Borasio GD (2007) Amyotrophic lateral sclerosis. Lancet 369:2031–2041. doi:10.1016/S0140-6736(07)60944-1
Moulignier A, Moulonguet A, Pialoux G, Rozenbaum W (2001) Reversible ALS-like disorder in HIV infection. Neurology 57(6):995–1001. doi:10.1212/WNL.57.6.995
Ou SH, Wu F, Harrich D, García-Martínez LF, Gaynor RB (1995) Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol 69(6):3584–3596
Pan YW, Storm DR, Xia Z (2013) Role of adult neurogenesis in hippocampus-dependent memory, contextual fear extinction and remote contextual memory: new insights from ERK5 MAP kinase. Neurobiol Learn Mem 105:81–92. doi:10.1016/j.nlm.2013.07.011
Philips T, Robberecht W (2011) Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol 10(3):253–263. doi:10.1016/S1474-4422(11)70015-1
Satterwhite E, Sonoki T, Willis TG, Harder L, Nowak R, Arriola EL, Liu H, Price HP, Gesk S, Steinemann D, Schlegelberger B, Oscier DG, Siebert R, Tucker PW, Dyer MJS (2001) The BCL11 gene family : involvement of BCL11A in lymphoid malignancies. Blood 98(12):3413–3420. doi:10.1182/blood.V98.12.3413
Simon R, Brylka H, Schwegler H, Venkataramanappa S, Andratschke J, Wiegreffe C, Liu P, Fuchs E, Jenkins NA, Copeland NG, Birchmeier C, Britsch S (2012) A dual function of Bcl11b/Ctip2 in hippocampal neurogenesis. EMBO J 31(13):2922–2936. doi:10.1038/emboj.2012.142
Tang B, Di Lena P, Schaffer L, Head SR, Baldi P, Thomas EA (2011) Genome-wide identification of Bcl11b gene targets reveals role in brain-derived neurotrophic factor signaling. PLoS One. doi:10.1371/journal.pone.0023691
Verma A, Berger JR (2006) ALS syndrome in patients with HIV-1 infection. J Neurol Sci 240(1–2):59–64. doi:10.1016/j.jns.2005.09.005
Zoccolella S, Beghi E, Palagano G, Fraddosio A, Guerra V, Samarelli V, Lepore V, Simone IL, Lamberti P, Serlenga L, Logroscino G (2007) Riluzole and amyotrophic lateral sclerosis survival: a population-based study in southern Italy. Eur J Neurol 14(3):262–268. doi:10.1111/j.1468-1331.2006.01575.x
Zuccato C, Cattaneo E (2009) Brain-derived neurotrophic factor in neurodegenerative diseases. Nature Reviews. Neurology 5(6):311–322. doi:10.1038/nrneurol.2009.54
Acknowledgments
The authors acknowledge the generous support and funding of the Peter Duncan Neurosciences Research Unit for this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Lennon, M.J., Jones, S.P., Lovelace, M.D. et al. Bcl11b: A New Piece to the Complex Puzzle of Amyotrophic Lateral Sclerosis Neuropathogenesis?. Neurotox Res 29, 201–207 (2016). https://doi.org/10.1007/s12640-015-9573-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-015-9573-5