
Abstract
Cadmium (Cd), a widely used industrial metal, is extremely nephrotoxic and neurotoxic; however, its effects on the peripheral auditory system are poorly understood. To evaluate the ototoxicity of Cd, we treated cochlear organotypic cultures from postnatal day 3 rats with Cd concentrations from 10 to 500 μM for 24 or 48 h. Afterward, we evaluated the degree of damage to hair cells, auditory nerve fibers, and spiral ganglion neurons. Damage to the hair cells, auditory nerve fibers, and spiral ganglion neurons systematically increased in a dose and time-dependent manner. Exposure to Cd concentrations of 10 μM for 24 and 48 h resulted in minor inner and outer hair cell loss in the basal third of the cochlea. As Cd concentrations increased, toxicity spread toward the apex, also in a time-dependent manner. Treatment with 100 μM Cd for 48 h resulted in substantial (>30 %) hair cell loss over the entire cochlea. Cd was also toxic to auditory nerve fibers and spiral ganglion neurons; 100 μM of Cd for 24 h or 10 μM of Cd for 48 h resulted in considerable damage to auditory nerve fibers and spiral ganglion neurons. These findings are the first to demonstrate that Cd can cause significant lesions to peripheral auditory nerve fibers, spiral ganglion neurons, and sensory hair cells in organotypic cultures from postnatal cochleae.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cadmium (Cd), a heavy metal widely used in the production of batteries, solar panels, pigments, and plastic stabilizers (Basinger et al. 1987; Bertin and Averbeck 2006; Herber 1992; Huff et al. 2007), has been linked to a wide range of adverse medical conditions (Cartana et al. 1992; Goering and Klaassen 1983; Nemery 1990; Reeves and Rossow 1996; Sullivan et al. 1984; Swiergosz-Kowalewska 2001) including cancer, nephrotoxicity, bone disease, and hepatotoxicity (Nordberg et al. 2012; Prozialeck et al. 2007). Cd typically enters the body from contaminated food and water as well as inhalation of polluted air and cigarette smoke (Friberg 1983; Massadeh et al. 2005). This was exemplified by residents of the Jinzu river basin in Japan who developed nephrotoxicity and itai–itai, a painful joint, skeletal, and bone disease as a result of Cd water pollution (Inaba et al. 2005; Kobayashi et al. 2009; Nogawa et al. 1983; Tsuchiya 1976). Humans and laboratory animals can also develop severe and irreversible neurological disorder due to excess Cd intake (Fern et al. 1996; Hart et al. 1989; Klaassen and Wong 1982; Kumar et al. 1996; Lopez et al. 2003; O’Callaghan and Miller 1986; Okuda et al. 1997; Webster and Valois 1981; Wong and Klaassen 1982). Cd-induced neurotoxicity is thought to arises from increased production of reactive oxygen species, depletion of antioxidant enzymes, and inhibition of cellular energy production resulting in apoptosis (National Center for Health Statistics 1988, Almar et al. 1987; Bianucci et al. 2011; Cookson and Pentreath 1996; El-Naggar and El-Sheekh 1998; Helbig et al. 2008; Kang 1992; MacKinnon and Kapron 2010; Muller 1986; Ochi et al. 1988; Rai et al. 1990; Shimizu and Morita 1990; Singhal et al. 1987; Sura et al. 2006; Tynecka et al. 1998; Yang et al. 2007).
The half life of Cd is in excess of 15 years accounting for its harmful accumulation in a variety of tissues. The highest concentrations of Cd are observed in the kidney, which is considered to be the major organ for the Cd related toxicity (Nogawa et al. 1983; Hamada et al. 1991). Several studies have suggested that the kidney is very similar to the cochlear portion of the inner ear given that many drugs that are nephrotoxic and also ototoxic (Humes 1999; Walker et al. 1990). Although research on the ototoxic effects of Cd is sparse, there is, in fact, evidence suggesting that Cd is toxic to the inner ear and can lead to hearing loss. For example, rats treated with CdCl2 (15 ppm) for 30 days in drinking water were found to have not only significant nephrotoxicity but also hearing loss as well (Ozcaglar et al. 2001). Cd treatment caused a reduction in otoacoustic emissions, suggesting that damage occurred to outer hair cells (OHC) and/or the stria vascularis (Ozcaglar et al. 2001). The latency of wave I of the auditory brainstem response (ABR), which originates from the auditory nerve, was also greatly delayed, suggesting possible damage to the spiral ganglion neurons (SGN). Surprisingly, the latencies of waves III and V, which emanate from the auditory brainstem, did not change significantly, suggesting that the cochlea is more vulnerable to the toxic effects of Cd than neurons in the auditory brainstem. Unfortunately, this study failed to perform any experimental verification of the suggested cochlear histopathologies responsible for these hearing defects. In contrast to chronic treatment, acute dosing of rats with Cd (5 mg/kg, i.p.) failed to produce any permanent hearing loss 7 day post-treatment (Whitworth et al. 1999); however, hearing and hair cell loss were observed when Cd was co-administered with furosemide, a diuretic that disrupts the blood-labyrinth, which by itself did not cause hearing impairment.
Thus, the existing literature, though limited, suggests that Cd has the potential to cause permanent hearing loss. It is unclear, however, as to which cells in the cochlea are preferentially damaged by Cd and whether cell death occurs predominantly by necrosis or apoptosis. To address these issues, we treated postnatal day 3 cochlear organotypic cultures with increasing concentrations of Cd to determine which concentrations were toxic to the cochlear hair cells, support cells, and SGN and to investigate some of the mechanisms by which Cd exerts its ototoxic effects.
Materials and Methods
Cochlear Organotypic Cultures
Procedures for preparing rat cochlear organotypic cultures have been described in previous publications (Ding et al. 2002, 2009b, 2011a; Qi et al. 2008; Wei et al. 2010). In brief, postnatal day 3 rat pups (SASCO Sprague–Dawley, Charles River Laboratory Inc.) were decapitated, and their cochleae were carefully removed. The whole cochlear basilar membrane containing the organ of Corti and SGN were dissected out in Hank’s Balanced Salt Solution and placed in a culture dish coated with rat tail collagen. Serum-free medium was added to the culture dish, and the cochlear explants were subsequently incubated at 37 °C in 5 % CO2 overnight. To determine the effect of Cd on cochlear damage, explants were cultured in fresh medium containing various concentrations of the metal for 24 or 48 h.
Histology
At the end of the incubation period, cochlear explants were fixed with 4 % formalin in phosphate buffered saline (PBS) for 1 h. Specimens were rinsed in 0.01 M PBS, incubated overnight (4 °C) in a solution containing of mouse anti-neurofilament 200 kD antibody, Triton X-100 (10 %), normal goat serum, and 0.01 M PBS. Specimens were subsequently rinsed with 0.01 M PBS, and then immersed for 1 h in a solution containing secondary antibody conjugated with Alexa Fluor 555 dissolved in 5 % normal goat serum, 1 % Triton X-100 in 0.01 M PBS. To label the stereocilia and the cuticular plate of cochlear hair cells, specimens were labeled with Alexa Fluor 488-phalloidin in PBS for 40 min. Afterward, specimens were mounted on glass slides in glycerin, coverslipped, and examined using a confocal microscope.
TUNEL Staining
Cochlear explants were cultured for 24 h in the presence or absence of 500 μM Cd. After fixation, specimens were fixed with 10 % formalin and processed for terminal deoxynucleotidyl transferase dUTP end-labeling (TUNEL) staining using a TUNEL Assay Kit (Invitrogen) according to the manufacturer’s protocol. Cochlear cultures were incubated in DNA-labeling solution at 37 °C for 4–8 h. After washing with rinsing buffer, specimens were incubated with antibody (5.0 μl of Alexa Fluor 488 dye-labeled anti-BrdU antibody and 95 μl rinsing buffer) at room temperature for 1 h, as previously described (Qi et al. 2008). Afterward, specimens were stained with an antibody against neurofilament 200 kD and secondary antibody (see above) to label SGN (Ding et al. 2007, 2011b; Wei et al. 2010). The nuclei in the cochlear cultures were also labeled with To-Pro-3 solution (1 mM To-Pro-3 in 0.75 μl DMSO dissolved in 1 ml H2O) at room temperature for about 30 min, then rinsed with 0.01 M PBS, and mounted on glass slides in glycerin.
Cochleograms
To quantify hair cell loss induced by Cd, cochlear cultures were examined under a fluorescent microscope (Zeiss Axioskop 400X) equipped with appropriate filters for detection of Alexa Fluor 488 fluorescence (absorption 488 nm, emission 520 nm). The numbers of missing IHC and OHC were counted in consecutive 0.24 mm segments along the entire length of the cochlea. Hair cells were counted as missing, if the stereocilia and the cuticular plate were absent. Using lab norms of hair cell counts from normal SASCO Sprague–Dawley rats, a cochleogram was constructed showing the percent IHC and OHC loss in 20 % intervals as a function of the percent distance from the apex of the cochlea. Hair cell losses from individual cochleograms were averaged to generate a mean (±SEM) cochleogram for each condition using custom software (Ding et al. 2009a, 2011a; Qi et al. 2008; Wei et al. 2010).
Confocal Microscopy
Specimens were examined under a confocal microscope (Zeiss LSM-510 meta, step size 0.5 μm per slice, approximately 10–20 slices) with appropriate filters for detection of Alexa Fluor 555 (absorption 555 nm, emission 565 nm), Alexa Fluor 488 (excitation 488 nm, emission 520 nm), or To-Pro-3 (absorption 642 nm, emission 661 nm). Specimens were processed with a Zeiss LSM Image Examiner (Zeiss LSM-510, step size 0.5 μm per slice, 20–50 slices). Images from approximately 10–20 slices were typically merged into a single plane using the Zeiss LSM Image Examiner (version: 4,0,0,91). Adobe Photoshop (version 5.5) was used for additional processing of photomicrographs as described previously (Ding et al. 2007, 2010, 2011a; Qi et al. 2008; Wei et al. 2010; Yu et al. 2011).
To quantify shrinkage of SGN soma induced by Cd treatment, the sizes of 150 SGN soma (6 cultures per condition; 25 SGN per culture) were measured from samples taken from the upper middle turn of the cochlea 24 h after being cultured without (control) or with 10, 100, or 500 μM Cd. Soma area was measured using Confocal LSM Image Examiner software as described previously (Wei et al. 2010). Briefly, multiple layers of SGN images were selected and merged into a single layer in which the SGNs with largest cross-sectional area were measured. To avoid repeated measurement of the same SGN, an interval of 20 μm was interposed between each of the merged layers. A polygon was drawn around the perimeter of the cell body of each well-defined SGN, and the enclosed area was calculated automatically by the Zeiss LSM Image Examiner software (version: 4,0,0,91). Statistical analyses were performed with GraphPad Prism program (version 5.01).
Results
Hair Cell Loss
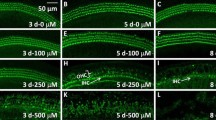
Initial studies were performed to examine the effects of Cd on hair cell viability. For these studies, cochlear cultures were labeled with Alexa Fluor 488-phalloidin in order to visualize the stereocilia bundle and the cuticular plate of the hair cells in both control and Cd-treated samples. Figure 1 shows a series of representative photomicrographs taken from the upper middle turn (~20 % of the distance from the apex) of the cochlea. Three orderly rows of OHC and one row of IHC were present in normal cochleas cultured for 24 h (Fig. 1a) or 48 h (Fig. 1e). Cochlear cultures treated with 10 μM Cd for 24 h (Fig. 1b) were similar to those of untreated controls. Cultures treated for 24 h with 100 μM Cd (Fig. 1c) showed little evidence of actual hair cells loss; however, the rows of OHC were slightly distorted due to inward and outward radial shifts of OHC (Fig. 1c, arrows). Treatment with 10 μM Cd for 48 h also resulted in severe distortion of the cochlear architecture, but displayed little hair cell loss (Fig. 1f). In some areas, the OHC shifted outward and away from the IHC, while in other regions the OHC shifted inward toward the IHC; these radial shifts resulted in rows of hair cells with a scalloped profile (Fig. 1f). When the dose of Cd was increased to 100 μM and treatment duration was 48 h, some hair cell rows disappeared and the rows became more distorted (Fig. 1g). A dose of 500 μM for 48 h destroyed nearly all the hair cells near the upper middle turn of the cochlea (Fig. 1h). Interestingly, when the treatment duration was decreased to 24 h, the 500 μM dose of Cd caused minimal hair cell loss, although considerable distortion of the hair cell rows was evident.

Representative photomicrographs of cochlear surface preparations labeled with Alexa Fluor 488-phalloidin taken from the upper middle turn of the cochlea approximately 20 % of the distance from the apex. Photomicrographs shown for normal controls (CNT) cultured for 24 h (a) or 48 h (e) and from cochleas treated for 24 h with 10–500 mM CdCl2 (b–d) or 48 h with 10–500 mM CdCl2 (f–h). Bracket in a shows 3 orderly rows of OHC; arrow shows the location of a single row of IHC. Note wavy row of OHC (dashed arrows) after 24 h treatment with 100 mM CD and highly disorganized rows of OHC after 48 h treatment with 10 mM Cd. Dashed arrows show inward and outward radial shift of OHC (f). Scale bar shown in lower right of (h)
To quantify the toxic effects of Cd on sensory hair cells, we counted the numbers of missing IHC and OHC along the entire length of the sensory epithelium and computed a mean cochleogram showing the percentage of missing hair cells as a function of percent distance from the apex for each experimental condition. Little or no hair cell loss was seen in normal control cochleas cultured for 24 h except for small IHC and OHC losses that increased from less than 10 % to approximately 30 % in the region 75–100 % of the distance from the apex (Fig. 2a). Control cochleas cultured for 48 h showed slightly greater loss of IHC and OHC 80–100 % distance from the apex plus less than 10 % OHC loss 0–10 % distance from the apex (Fig. 2e). These lesions are most likely the result of mechanical damage that occurred during the dissection and preparation of the cultures as noted previously (Wu et al. 2011a). Cultures treated for 24 h with 10 μM Cd had greater IHC and OHC loss in the basal third of the cochlea (Fig. 2b) than controls cultured for 24 h (Fig. 2a). As the Cd concentration increased from 10 to 100 μM, IHC and OHC losses increased and expanded over the basal half (50–100 %) of the cochlea (Fig. 2c). The 500 μM dose for 24 h resulted in extensive IHC and OHC over the entire cochlea; the losses decreased from 100 % near the base of the cochlea to approximately 40 % IHC and 60 % OHC loss in the apical third of the cochlea (Fig. 2d). Treatment for 48 h with 10 μM Cd resulted in approximately 70 % IHC and OHC loss in the extreme base of the cochlea to roughly 10 % IHC and OHC loss in the middle of the cochlea (Fig. 2f); these losses were considerably greater than those seen in the 48 h controls (Fig. 1e). With 100 μM Cd, the IHC and OHC lesion expanded all the way to the apex; nearly all of the IHC and OHC were missing in the extreme base of the cochlea, whereas roughly 30 % of the IHC and 40 % of the OHC were missing between 20 and 80 % of the distance from the apex of the cochlea (Fig. 2g). Treatment with 500 μM Cd for 48 h resulted in 100 % loss of IHC and OHC over the basal half of the cochlea; OHC and IHC losses then declined to roughly 50 and 40 %, respectively, near the apex of the cochlea (Fig. 2h). The Cd-induced hair cell lesions generally progressed from base to apex as the dose increased; although the OHC lesions tended to be slightly greater than IHC, the differences were relatively small.

Mean (±SEM) cochleograms (n = 6/condition) showing the percentage of missing IHC and OHC in 20 % intervals as function of percent distance from the apex. Data shown for normal controls maintained in culture for 24 h (a) or 48 h (e). b, c, and d show the mean cochleograms obtained from cultures treated for 24 h with 10, 100, or 500 μM of CdCl2. f, g, and h show the mean cochleograms obtained from cultures treated for 48 h with 10, 100, or 500 μM CdCl2. Approximately 23 % of IHC and OHC were missing at the extreme base in normal control cultured for 24 or 48 h presumably due to mechanical damage during the sample preparations
Auditory Nerve Fibers and Spiral Ganglion Damage
Studies were also performed to determine the effects of Cd on both the auditory nerve fibers and spiral ganglion. For these experiments, specimens were labeled with Alexa-Fluor 488-phalloidin to identify hair cells and neurofilament-Alexa-Fluor 555 antibody to visualize SGN and auditory nerve fibers (NF). Representative confocal images from the upper middle turn of control and Cd-treated cochleas cultured for 24 h are shown in Fig. 3a–d. In control samples, thick fascicles of NF can be seen emanating from the SGN and projecting out radially to the three rows of OHC and one row of IHC (Fig. 3a). The NF and SGN in cultures treated with 10 μM Cd (Fig. 3b) appeared similar to controls (Fig. 3b). As the dose of Cd increased from 100 to 500 μM Cd (Fig. 2c–d), the NF fascicles began to thin out and fragment, whereas the soma of the SGN began to shrink and disappear. The dose-dependent damage to the NF increased at approximately the same rate as the damage to hair cells. The NF and SGN in untreated controls cultured for 48 h (Fig. 3e) appeared normal and similar to those cultured 24 h. However, the NF fascicles started to thin out (Fig. 3f) after 48 h treatment with 10 μM Cd. NF shrinkage and fragmentation and SGN shrinkage increased rapidly as the dose of Cd increased from 100 to 500 μM (Fig. 3f–h). Nearly all the hair cells, NF, and SGN were destroyed after 48 h treatment with 500 μM Cd.

Confocal images of control and cadmium-treated cochlear explants cultured for 24 h (a–d) or 48 h (e–h). Hair cells (HC) labeled with Alexa-Fluor 488 phalloidin (green, yellow label). Auditory nerve fibers (NF) and spiral ganglion neurons (SGN) labeled with neurofilament 200 kD-Alexa-Fluor 555 (red). In controls (CNT) cultured for 24 h (a) or 48 h (e), thick fascicles of NF project out radially from SGN soma toward the HC. Cochlear explants cultured with increasing concentrations of cadmium for 24 h (b–d) or 48 h (f–h). Note the dose-dependent loss of NF and SGN beginning with 100 μM after 24 h treatment and beginning with 10 μM after 48 h treatment (Color figure online)
The higher magnification confocal images displayed in Fig. 4 further illustrate the changes in the size and shape of the soma and nucleus of SGN when cultured for 24 h with increasing concentrations of Cd. Soma and nuclear shrinkage was apparent in some SGN at the lowest concentration, 10 μM, whereas at the highest concentration, 500 μM, most of the soma and nuclei were severely shrunken and condensed, anatomical hallmarks of apoptosis. In addition, many SGN were missing. To quantify the shrinkage of SGN soma, we measured the cross-sectional area of 25 SGN soma located in the upper middle turn of each sample (n = 6 samples per condition) after 24 h in culture. Measurements were obtained from untreated controls and cultures treated with 10, 100, or 500 μM Cd. The mean size of the SGN in normal controls was 220.2 μm2 (SEM 2.24 μm2; Fig. 5). Soma cross-sectional area decreased in a dose-dependent manner from 207 μm2 (SEM 1.96 μm2) at 10 μM, 157 μm2 at 100 μM (SEM 1.99 μm2), and 90.8 μm2 at 500 μM (SEM 2.52 μm2). A one-way analysis of variance revealed a main effect of Cd dose (F = 720.5, 3, 596 df, p < 0.0056), and a Tukey post hoc analysis showed a significant difference (p < 0.05) between all treatment groups as indicated by the arrows in Fig. 5.

Representative confocal images of SGN cultured for 24 h without and with Cd. SGN labeled with neurofilament 200 kD-Alexa-Fluor 555 (red). a SGN in controls (CNT) have a large soma (white arrow) and round nucleolus. Some thin nerve fibers (NF, yellow arrowhead) pass by the SGN. (b–d) Soma and nuclei of SGN became progressively smaller and more condensed as the dose of Cd increased from 10 to 500 μM. Note the loss of SGN and NF in cultures treated with 500 μM Cd. Scale bar shown in a (Color figure online)

Mean (n = 6, ±SEM) area of SGN soma in control and Cd (10–500 μM)-treated cochleas cultured for 24 h. Arrows indicate conditions that were significantly different from one another (p < 0.05, Tukey post hoc analysis)
Cd-Induced Apoptosis
To determine if Cd-induced cochlear degeneration was mediated by apoptosis, cochlear explants treated with or without 500 μM Cd for 48 h were evaluated with TUNEL staining to identify DNA fragmentation. Results of these experiments obtained with untreated control cochlear explants revealed no TUNEL-positive labeling in IHC, OHC, or SGN (Fig. 6a, c). In contrast, many TUNEL-positive cells were seen in the hair cell region (Fig. 6b) and in the SGN region (Fig. 6d) 48 h after 500 μM Cd treatment.

a–b Representative confocal images of cochlear cultures stained with To-Pro-3 (blue fluorescence of To-Pro-3 converted to red for optimal clarity) and TUNEL Assay Kit (green). a Control cochlea cultured for 48 h; note the robust To-Pro-3 in nuclei of IHC and OHC and the absence of TUNEL labeling. b Cochlea cultured for 48 h with 500 μM Cd; note TUNEL labeling (green or yellow, white arrowheads) in region normally occupied by hair cells (HC). c–d Representative confocal images of SGN; specimens labeled with neurofilament 200 kD-Alexa-Fluor 555 (red) and TUNEL Assay Kit (green/yellow). c SGN in normal controls cultured for 48 h have a large round soma and large nucleolus (arrows); note the absence of TUNEL labeling in controls. d SGN treated with 500 μM Cd for 48 h; note TUNEL labeling (white arrowhead) in SGN with shrunken soma (white arrowheads) and condensed nuclei (Color figure online)
Discussion
Cd is an environmental contaminant most commonly produced from industrial pollution during smelting and refining of metals and manufacturing of batteries, alloys, and pigments used in production of inks, dyes, paints, enamels, and plastics. Significant exposure to Cd can also result from its high content in cigarette smoke. Cd is on the EPA National Emission Standards for Hazardous Air Pollutants (NESHAP) list of 189 hazardous air pollutants and is listed as one of 33 hazardous air pollutants that present the greatest threat to public health in urban areas. Chronic exposure causes Cd to accumulate in the body as the biological half life is estimated to be at least 15 years (Dickel et al. 2002; Viaene et al. 1999, 2000). Since Cd absorption in the body is relatively low, when testing the toxic effects of Cd in experimental settings, it is difficult to fully imitate the cumulative process that occurs in animals with real exposure in nature. Although, not without its limitations, it is possible to shorten the long period of exposure to heavy metals in vitro by increasing their operative concentrations in the culture dish (Ding et al. 2011a; Kim et al. 2010; Leoni et al. 2002; Prins et al. 2010; Schmid et al. 1983; Waters et al. 1975; Wu et al. 2011b). In the current study, cochlear explants were treated with Cd at concentrations ranging from 10 to 500 μM. These were equivalent to 1.83 μg per milliliter in 10 μM Cd and 92 μg per milliliter in 500 μM Cd in culture medium, respectively. These were approximately 100–5,000 times higher than the safety standards (15 μg/L) for workers according to German law (Godt et al. 2006).
Numerous studies have demonstrated that Cd can produce severe neurotoxic manifestations especially in the central and peripheral nervous system in neonatal animals, which is believed to be caused by amplified sensitivity in the developing brain (Antonio et al. 2003; Rigon et al. 2008; Viaene et al. 1999). Although the ototoxic effects of Cd have previously been described in the literature (Agirdir et al. 2002; Kim et al. 2008, 2009), detailed information as to the mechanism of the actions of Cd on the different cells within the cochlea has not previously been described. In the current study, we provide the first evidence that Cd-induced ototoxicity involves structural degeneration of HCs, peripheral auditory nerve fibers and SGN.
As indicated by the results presented in this paper, Cd-induced damage to hair cells, auditory nerve fibers and SGN systematically increased in a dose and time-dependent manner. Exposure to Cd concentrations of 10 μM for 24 and 48 h resulted in minor IHC and OHC loss in the basal third of the cochlea. As Cd concentrations were increased toxicity spread toward the apex, also in a time-dependent manner. Treatment with 100 μM Cd for 48 h resulted in substantial hair cell loss over the entire cochlea as well as toxicity to auditory nerve fibers and SGN. Higher concentrations of Cd led to further degeneration of hair cells as well as breakdown of the peripheral auditory nerve fibers and the soma of SGN.
Loss of IHC and OHC followed a consistent pattern of damage that begins at the base of the cochlea and progresses toward the apex. This is similar to most ototoxic drugs, such as aminoglycoside antibiotics, antitumor platinum drugs, and other toxic reagents (Ding et al. 2011b; Ding and Salvi 2004, 2007; Li et al. 2011; Qi et al. 2008). These findings are consistent with previous studies showing that Cd causes hearing loss and cochlear HC loss in experimental animal models in vivo (Agirdir et al. 2002; Ozcaglar et al. 2001; Prasher 2009; Whitworth et al. 1999).
Previous studies indicated that Cd can accumulate in the inner ear following Cd treatment causing selective prolongation of latency in ABR wave I, but failing to change the latencies of either wave III or V (Ozcaglar et al. 2001). These findings suggest that cochlear HCs as well as peripheral auditory neurons are more vulnerable to Cd than cells within the central auditory system. Our results demonstrated a concentration and duration-dependent damage effect on peripheral auditory nerve fibers and SGNs in rat cochlear organotypic cultures exposed to Cd. Since the ABR wave I is derived from the auditory nerve and SGN that contact cochlear IHC, the current findings are consistent with prior results demonstrating Cd-induced dysfunction in the auditory periphery in vivo (Ozcaglar et al. 2001).
Cd is known to disrupt E-cadherin mediated adhesion in pig kidney epithelial, LLC-PK1, and MDCK cells in vitro (Prozialeck 2000; Prozialeck and Lamar 1997; Prozialeck and Niewenhuis 1991) as well as to alter the pattern of N-cadherin localization in the kidneys of rats sub-chronically exposed to Cd (Prozialeck et al. 2003). In both in vitro and in vivo studies, the Cd doses and durations of exposure employed failed to produce overt signs of necrotic or apoptotic cell death (Prozialeck 2000; Prozialeck et al. 2003). The implications of these findings are that cadherins are the potential targets of Cd-induced injury. Relevant to the current finding, mutations in the genes encoding cadherin 23 and protocadherin 15 have been shown to be necessary for normal hair cell mechanotransduction (Alagramam et al. 2011; Kazmierczak et al. 2007), and that mutations of these genes can lead to hearing loss (Kane et al. 2012; Liu et al. 2012). Importantly, our results show that 48 h treatment with a Cd concentration as low as 10 μM was sufficient to disrupt the organization of hair cells causing severe distortion of the rows of hair cells and cochlear architecture, but little hair cell loss. This suggests that the initial effect of Cd may not be hair cell degeneration, but rather disruption of the cadherin intercellular junctions between hair cells and supporting cells. This raises the possibility that hair cell death is induced by the detachment of cells from the surrounding extracellular matrix, a form of cell death referred to as anoikis (Loza-Coll et al. 2005; Weng et al. 2002).
Although Cd toxicity is implicated in a diverse array of pathological conditions, the precise mechanism by which it produces neurotoxicity and ototoxicity is poorly understood. The general consensus is that cell death is typically mediated by apoptosis, although necrosis has also been proposed to be a contributing factor (Prozialeck et al. 2009). Necrosis, can induce an inflammatory response in vivo (Hughes and Gobe 2007), and this would likely exacerbate any oxidative stress caused by Cd alone. Cd may also induce autophagy (Wang et al. 2008) which allows for the digestion of dysfunctional organelles within lysosomal autophagic vacuoles. Autophagy results in the removal of dysfunctional mitochondria which potentially can reduce excessive formation of ROS, but, may, also cause digestion of a sufficient number of mitochondria such that cell viability is compromised (Sansanwal et al. 2010). In the experiments reported here, TUNEL staining and cellular histopathologies demonstrated that Cd-induced damage to IHC, OHC, and SGN that resulted in nuclear condensations and/or fragmentations, morphologic features of apoptosis. These findings are consistent with numerous other studies demonstrating that Cd cell death is mediated by apoptosis (Chabicovsky et al. 2004; Chao and Yang 2001; Chatterjee et al. 2009; Chuang et al. 2000; Coutant et al. 2006; Dong et al. 2001; Fernandez et al. 2003; Galan et al. 2000; Joseph 2009; Jung et al. 2008; Kim et al. 2000; Kondoh et al. 2002; Lasfer et al. 2008; Li et al. 2003; Mao et al. 2007; Pathak and Khandelwal 2006; Poliandri et al. 2003; Shih et al. 2004; Xu et al. 1996).
In conclusion, our results demonstrate for the first time both a time and concentration-dependent increase in the toxic effects of Cd on IHC, OHC, auditory nerve fibers, and SGN in organotypic cultures from postnatal day 3 cochleae. Cell death most likely was mediated by apoptosis as indicted by TUNEL staining, indicative of DNA strand breaks, in IHC, OHC, and SGN; these changes were associated with soma shrinkage and nuclear condensation and fragmentation.
References
Agirdir BV, Bilgen I, Dinc O, Ozcaglar HU, Fisenk F, Turhan M et al (2002) Effect of zinc ion on cadmium-induced auditory changes. Biol Trace Elem Res 88:153–163
Alagramam KN, Goodyear RJ, Geng R, Furness DN, van Aken AF, Marcotti W et al (2011) Mutations in protocadherin 15 and cadherin 23 affect tip links and mechanotransduction in Mammalian sensory hair cells. PLoS ONE 6:e19183
Almar MM, Diaz-Mayans J, Romero FJ (1987) Glutathione content and GSH S-transferase activity in midgut gland of Procambarus clarkii. Sex differences, the effect of fasting, and their implications in cadmium toxicity. Comp Biochem Physiol C 87:433–435
Antonio MT, Corredor L, Leret ML (2003) Study of the activity of several brain enzymes like markers of the neurotoxicity induced by perinatal exposure to lead and/or cadmium. Toxicol Lett 143:331–340
Basinger MA, Jones MM, Craft WD, Walker EM Jr, Sanders MM (1987) Chelating-agent suppression of cadmium-induced hepatotoxicity. J Toxicol Environ Health 22:261–271
Bertin G, Averbeck D (2006) Cadmium: cellular effects, modifications of biomolecules, modulation of DNA repair and genotoxic consequences (a review). Biochimie 88:1549–1559
Bianucci E, Fabra A, Castro S (2011) Involvement of glutathione and enzymatic defense system against cadmium toxicity in Bradyrhizobium sp. strains (peanut symbionts). Biometals 25:23
Cartana J, Romeu A, Arola L (1992) Effects of copper, cadmium and nickel on liver and kidney glutathione redox cycle of rats (Rattus sp.). Comp Biochem Physiol C 101:209–213
Chabicovsky M, Klepal W, Dallinger R (2004) Mechanisms of cadmium toxicity in terrestrial pulmonates: programmed cell death and metallothionein overload. Environ Toxicol Chem 23:648–655
Chao JI, Yang JL (2001) Opposite roles of ERK and p38 mitogen-activated protein kinases in cadmium-induced genotoxicity and mitotic arrest. Chem Res Toxicol 14:1193–1202
Chatterjee S, Kundu S, Sengupta S, Bhattacharyya A (2009) Divergence to apoptosis from ROS induced cell cycle arrest: effect of cadmium. Mutat Res 663:22–31
Chuang SM, Wang IC, Yang JL (2000) Roles of JNK, p38 and ERK mitogen-activated protein kinases in the growth inhibition and apoptosis induced by cadmium. Carcinogenesis 21:1423–1432
Cookson MR, Pentreath VW (1996) Protective roles of glutathione in the toxicity of mercury and cadmium compounds to C6 glioma cells. Toxicol In Vitro 10:257–264
Coutant A, Lebeau J, Bidon-Wagner N, Levalois C, Lectard B, Chevillard S (2006) Cadmium-induced apoptosis in lymphoblastoid cell line: involvement of caspase-dependent and -independent pathways. Biochimie 88:1815–1822
Dickel H, Kuss O, Schmidt A, Diepgen TL (2002) Occupational relevance of positive standard patch-test results in employed persons with an initial report of an occupational skin disease. Int Arch Occup Environ Health 75:423–434
Ding D, Salvi R (2004) Review of cellular changes in the cochlea due to aminoglycoside antibiotics. Volta Rev 105:407–438
Ding D, Salvi R (2007) Experiences in ototoxic investigations in aminoglycoside antibiotics. Chin J Otol 5:125–131
Ding D, Stracher A, Salvi RJ (2002) Leupeptin protects cochlear and vestibular hair cells from gentamicin ototoxicity. Hear Res 164:115–126
Ding D, Jiang H, Wang P, Salvi R (2007) Cell death after co-administration of cisplatin and ethacrynic acid. Hear Res 226:129–139
Ding D, Qi W, Yu D, Jiang H, Salvi R (2009a) Ototoxic effects of mefloquine in cochlear organotypic cultures. J Otol 4:29–38
Ding D, Wang P, Jiang H, Coling D, Salvi R (2009b) Gene expression in cisplatin ototoxicity and protection with p53 inhibitor. J Otol 4:15–24
Ding D, Jiang H, Salvi RJ (2010) Mechanisms of rapid sensory hair-cell death following co-administration of gentamicin and ethacrynic acid. Hear Res 259:16–23
Ding D, He J, Allman BL, Yu D, Jiang H, Seigel GM et al (2011a) Cisplatin ototoxicity in rat cochlear organotypic cultures. Hear Res 282:196–203
Ding D, Roth J, Salvi R (2011b) Manganese is toxic to spiral ganglion neurons and hair cells in vitro. Neurotoxicology 32:233–241
Dong S, Shen HM, Ong CN (2001) Cadmium-induced apoptosis and phenotypic changes in mouse thymocytes. Mol Cell Biochem 222:11–20
El-Naggar AH, El-Sheekh MM (1998) Abolishing cadmium toxicity in Chlorella vulgaris by ascorbic acid, calcium, glucose and reduced glutathione. Environ Pollut 101:169–174
Fern R, Black JA, Ransom BR, Waxman SG (1996) Cd(2+)-induced injury in CNS white matter. J Neurophysiol 76:3264–3273
Fernandez EL, Gustafson AL, Andersson M, Hellman B, Dencker L (2003) Cadmium-induced changes in apoptotic gene expression levels and DNA damage in mouse embryos are blocked by zinc. Toxicol Sci 76:162–170
Friberg L (1983) Cadmium. Annu Rev Public Health 4:367–373
Galan A, Garcia-Bermejo ML, Troyano A, Vilaboa NE, de Blas E, Kazanietz MG et al (2000) Stimulation of p38 mitogen-activated protein kinase is an early regulatory event for the cadmium-induced apoptosis in human promonocytic cells. J Biol Chem 275:11418–11424
Godt J, Scheidig F, Grosse-Siestrup C, Esche V, Brandenburg P, Reich A et al (2006) The toxicity of cadmium and resulting hazards for human health. J Occup Med Toxicol 1:22
Goering PL, Klaassen CD (1983) Altered subcellular distribution of cadmium following cadmium pretreatment: possible mechanism of tolerance to cadmium-induced lethality. Toxicol Appl Pharmacol 70:195–203
Hamada T, Nakano S, Iwai S, Tanimoto A, Ariyoshi K, Koide O (1991) Pathological study on beagles after long-term oral administration of cadmium. Toxicol Pathol 19:138–47.
Hart RP, Rose CS, Hamer RM (1989) Neuropsychological effects of occupational exposure to cadmium. J Clin Exp Neuropsychol 11:933–943
Helbig K, Grosse C, Nies DH (2008) Cadmium toxicity in glutathione mutants of Escherichia coli. J Bacteriol 190:5439–5454
Herber RF (1992) The World Health Organization study on health effects of exposure to cadmium: morbidity studies. IARC Sci Publ 118:347–358
Huff J, Lunn RM, Waalkes MP, Tomatis L, Infante PF (2007) Cadmium-induced cancers in animals and in humans. Int J Occup Environ Health 13:202–212
Hughes J, Gobe G (2007) Identification and quantification of apoptosis in the kidney using morphology, biochemical and molecular markers. Nephrology (Carlton) 12:452–458
Humes HD (1999) Insights into ototoxicity. Analogies to nephrotoxicity. Ann N Y Acad Sci 884:15–18
Inaba T, Kobayashi E, Suwazono Y, Uetani M, Oishi M, Nakagawa H et al (2005) Estimation of cumulative cadmium intake causing Itai–itai disease. Toxicol Lett 159:192–201
Joseph P (2009) Mechanisms of cadmium carcinogenesis. Toxicol Appl Pharmacol 238:272–279
Jung YS, Jeong EM, Park EK, Kim YM, Sohn S, Lee SH et al (2008) Cadmium induces apoptotic cell death through p38 MAPK in brain microvessel endothelial cells. Eur J Pharmacol 578:11–18
Kane KL, Longo-Guess CM, Gagnon LH, Ding D, Salvi RJ, Johnson KR (2012) Genetic background effects on age-related hearing loss associated with Cdh23 variants in mice. Hear Res 283:80–88
Kang YJ (1992) Exogenous glutathione decreases cellular cadmium uptake and toxicity. Drug Metab Dispos 20:714–718
Kazmierczak P, Sakaguchi H, Tokita J, Wilson-Kubalek EM, Milligan RA, Muller U et al (2007) Cadherin 23 and protocadherin 15 interact to form tip-link filaments in sensory hair cells. Nature 449:87–91
Kim MS, Kim BJ, Woo HN, Kim KW, Kim KB, Kim IK et al (2000) Cadmium induces caspase-mediated cell death: suppression by Bcl-2. Toxicology 145:27–37
Kim SJ, Jeong HJ, Myung NY, Kim MC, Lee JH, So HS et al (2008) The protective mechanism of antioxidants in cadmium-induced ototoxicity in vitro and in vivo. Environ Health Perspect 116:854–862
Kim SJ, Shin BG, Choi IY, Kim DH, Kim MC, Myung NY et al (2009) Hwanggunchungyitang prevents cadmium-induced ototoxicity through suppression of the activation of caspase-9 and extracellular signal-related kinase in auditory HEI-OC1 cells. Biol Pharm Bull 32:213–219
Kim K, Fujimoto VY, Parsons PJ, Steuerwald AJ, Browne RW, Bloom MS (2010) Recent cadmium exposure among male partners may affect oocyte fertilization during in vitro fertilization (IVF). J Assist Reprod Genet 27:463–468
Klaassen CD, Wong KL (1982) Cadmium toxicity in the newborn rat. Can J Physiol Pharmacol 60:1027–1036
Kobayashi E, Suwazono Y, Dochi M, Honda R, Kido T, Nakagawa H (2009) Influence of drinking and/or cooking with Jinzu River water on the development of Itai–itai disease. Biol Trace Elem Res 129:46–57
Kondoh M, Araragi S, Sato K, Higashimoto M, Takiguchi M, Sato M (2002) Cadmium induces apoptosis partly via caspase-9 activation in HL-60 cells. Toxicology 170:111–117
Kumar R, Agarwal AK, Seth PK (1996) Oxidative stress-mediated neurotoxicity of cadmium. Toxicol Lett 89:65–69
Lasfer M, Vadrot N, Aoudjehane L, Conti F, Bringuier AF, Feldmann G et al (2008) Cadmium induces mitochondria-dependent apoptosis of normal human hepatocytes. Cell Biol Toxicol 24:55–62
Leoni G, Bogliolo L, Deiana G, Berlinguer F, Rosati I, Pintus PP et al (2002) Influence of cadmium exposure on in vitro ovine gamete dysfunction. Reprod Toxicol 16:371–377
Li M, Xia T, Jiang CS, Li LJ, Fu JL, Zhou ZC (2003) Cadmium directly induced the opening of membrane permeability pore of mitochondria which possibly involved in cadmium-triggered apoptosis. Toxicology 194:19–33
Li Y, Ding D, Jiang H, Fu Y, Salvi R (2011) Co-administration of cisplatin and furosemide causes rapid and massive loss of cochlear hair cells in mice. Neurotox Res 20:307–319
Liu S, Li S, Zhu H, Cheng S, Zheng QY (2012) A mutation in the cdh23 gene causes age-related hearing loss in Cdh23(nmf308/nmf308) mice. Gene 499:309–317
Lopez E, Figueroa S, Oset-Gasque MJ, Gonzalez MP (2003) Apoptosis and necrosis: two distinct events induced by cadmium in cortical neurons in culture. Br J Pharmacol 138:901–911
Loza-Coll MA, Perera S, Shi W, Filmus J (2005) A transient increase in the activity of Src-family kinases induced by cell detachment delays anoikis of intestinal epithelial cells. Oncogene 24:1727–1737
MacKinnon Y, Kapron CM (2010) Reduction in cadmium-induced toxicity and c-Jun N-terminal kinase activation by glutathione in cultured mouse embryonic cells. Birth Defects Res A Clin Mol Teratol 88:707–714
Mao WP, Ye JL, Guan ZB, Zhao JM, Zhang C, Zhang NN et al (2007) Cadmium induces apoptosis in human embryonic kidney (HEK) 293 cells by caspase-dependent and -independent pathways acting on mitochondria. Toxicol In Vitro 21:343–354
Massadeh AM, Alali FQ, Jaradat QM (2005) Determination of cadmium and lead in different cigarette brands in Jordan. Environ Monit Assess 104:163–170
Muller L (1986) Consequences of cadmium toxicity in rat hepatocytes: effects of cadmium on the glutathione-peroxidase system. Toxicol Lett 30:259–265
National Center for Health Statistics (1988). Data from the National Health Interview Survey
Nemery B (1990) Metal toxicity and the respiratory tract. Eur Respir J 3:202–219
Nogawa K, Yamada Y, Honda R, Ishizaki M, Tsuritani I, Kawano S et al (1983) The relationship between itai–itai disease among inhabitants of the Jinzu River basin and cadmium in rice. Toxicol Lett 17:263–266
Nordberg G, Jin T, Wu X, Lu J, Chen L, Liang Y et al (2012) Kidney dysfunction and cadmium exposure-factors influencing dose-response relationships. J Trace Elem Med Biol 26:197–200
O'Callaghan JP, Miller DB (1986) Diethyldithiocarbamate increases distribution of cadmium to brain but prevents cadmium-induced neurotoxicity. Brain Res 370:354–358
Ochi T, Otsuka F, Takahashi K, Ohsawa M (1988) Glutathione and metallothioneins as cellular defense against cadmium toxicity in cultured Chinese hamster cells. Chem Biol Interact 65:1–14
Okuda B, Iwamoto Y, Tachibana H, Sugita M (1997) Parkinsonism after acute cadmium poisoning. Clin Neurol Neurosurg 99:263–265
Ozcaglar HU, Agirdir B, Dinc O, Turhan M, Kilincarslan S, Oner G (2001) Effects of cadmium on the hearing system. Acta Otolaryngol 121:393–397
Pathak N, Khandelwal S (2006) Modulation of cadmium induced alterations in murine thymocytes by piperine: oxidative stress, apoptosis, phenotyping and blastogenesis. Biochem Pharmacol 72:486–497
Poliandri AH, Cabilla JP, Velardez MO, Bodo CC, Duvilanski BH (2003) Cadmium induces apoptosis in anterior pituitary cells that can be reversed by treatment with antioxidants. Toxicol Appl Pharmacol 190:17–24
Prasher D (2009) Heavy metals and noise exposure: health effects. Noise Health 11:141–144
Prins JM, Brooks DM, Thompson CM, Lurie DI (2010) Chronic low-level Pb exposure during development decreases the expression of the voltage-dependent anion channel in auditory neurons of the brainstem. Neurotoxicology 31:662–673
Prozialeck WC (2000) Evidence that E-cadherin may be a target for cadmium toxicity in epithelial cells. Toxicol Appl Pharmacol 164:231–249
Prozialeck WC, Lamar PC (1997) Cadmium (Cd2+) disrupts E-cadherin-dependent cell–cell junctions in MDCK cells. In Vitro Cell Dev Biol Anim 33:516–526
Prozialeck WC, Niewenhuis RJ (1991) Cadmium (Cd2+) disrupts intercellular junctions and actin filaments in LLC-PK1 cells. Toxicol Appl Pharmacol 107:81–97
Prozialeck WC, Lamar PC, Lynch SM (2003) Cadmium alters the localization of N-cadherin, E-cadherin, and beta-catenin in the proximal tubule epithelium. Toxicol Appl Pharmacol 189:180–195
Prozialeck WC, Vaidya VS, Liu J, Waalkes MP, Edwards JR, Lamar PC et al (2007) Kidney injury molecule-1 is an early biomarker of cadmium nephrotoxicity. Kidney Int 72:985–993
Prozialeck WC, Edwards JR, Lamar PC, Liu J, Vaidya VS, Bonventre JV (2009) Expression of kidney injury molecule-1 (Kim-1) in relation to necrosis and apoptosis during the early stages of Cd-induced proximal tubule injury. Toxicol Appl Pharmacol 238:306–314
Qi W, Ding D, Salvi RJ (2008) Cytotoxic effects of dimethyl sulphoxide (DMSO) on cochlear organotypic cultures. Hear Res 236:52–60
Rai LC, Jensen TE, Rachlin JW (1990) A morphometric and X-ray energy dispersive approach to monitoring pH-altered cadmium toxicity in Anabaena flos-aquae. Arch Environ Contam Toxicol 19:479–487
Reeves PG, Rossow KL (1996) Zinc-and/or cadmium-induced intestinal metallothionein and copper metabolism in adult rats. NutrBiochem 7:128–134
Rigon AP, Cordova FM, Oliveira CS, Posser T, Costa AP, Silva IG et al (2008) Neurotoxicity of cadmium on immature hippocampus and a neuroprotective role for p38 MAPK. Neurotoxicology 29:727–734
Sansanwal P, Yen B, Gahl WA, Ma Y, Ying L, Wong LJ et al (2010) Mitochondrial autophagy promotes cellular injury in nephropathic cystinosis. J Am Soc Nephrol 21:272–283
Schmid BP, Hall JL, Goulding E, Fabro S, Dixon R (1983) In vitro exposure of male and female mice gametes to cadmium chloride during the fertilization process, and its effects on pregnancy outcome. Toxicol Appl Pharmacol 69:326–332
Shih CM, Ko WC, Wu JS, Wei YH, Wang LF, Chang EE et al (2004) Mediating of caspase-independent apoptosis by cadmium through the mitochondria-ROS pathway in MRC-5 fibroblasts. J Cell Biochem 91:384–397
Shimizu M, Morita S (1990) Effects of fasting on cadmium toxicity, glutathione metabolism, and metallothionein synthesis in rats. Toxicol Appl Pharmacol 103:28–39
Singhal RK, Anderson ME, Meister A (1987) Glutathione, a first line of defense against cadmium toxicity. FASEB J 1:220–223
Sullivan MF, Miller BM, Goebel JC (1984) Gastrointestinal absorption of metals (51Cr, 65Zn, 95mTc, 109Cd, 113Sn, 147Pm, and 238Pu) by rats and swine. Environ Res 35:439–453
Sura P, Ristic N, Bronowicka P, Wrobel M (2006) Cadmium toxicity related to cysteine metabolism and glutathione levels in frog Rana ridibunda tissues. Comp Biochem Physiol C 142:128–135
Swiergosz-Kowalewska R (2001) Cadmium distribution and toxicity in tissues of small rodents. Microsc Res Tech 55:208–222
Tsuchiya K (1976) Epidemiological studies on cadmium in the environment in Japan: etiology of itai–itai disease. Fed Proc 35:2412–2418
Tynecka Z, Malm A, Kosikowska U, Kot A (1998) Substrate-dependent cadmium toxicity affecting energy-linked K+/86Rb transport in Staphylococcus aureus. Folia Microbiol (Praha) 43:617–622
Viaene MK, Roels HA, Leenders J, De Groof M, Swerts LJ, Lison D et al (1999) Cadmium: a possible etiological factor in peripheral polyneuropathy. Neurotoxicology 20:7–16
Viaene MK, Masschelein R, Leenders J, De Groof M, Swerts LJ, Roels HA (2000) Neurobehavioural effects of occupational exposure to cadmium: a cross sectional epidemiological study. Occup Environ Med 57:19–27
Walker EM Jr, Fazekas-May MA, Bowen WR (1990) Nephrotoxic and ototoxic agents. Clin Lab Med 10:323–354
Wang SH, Shih YL, Ko WC, Wei YH, Shih CM (2008) Cadmium-induced autophagy and apoptosis are mediated by a calcium signaling pathway. Cell Mol Life Sci 65:3640–3652
Waters MD, Gardner DE, Aranyi C, Coffin DL (1975) Metal toxicity for rabbit alveolar macrophages in vitro. Environ Res 9:32–47
Webster WS, Valois AA (1981) The toxic effects of cadmium on the neonatal mouse CNS. J Neuropathol Exp Neurol 40:247–257
Wei L, Ding D, Salvi R (2010) Salicylate-induced degeneration of cochlea spiral ganglion neurons-apoptosis signaling. Neuroscience 168:288–299
Weng Z, Xin M, Pablo L, Grueneberg D, Hagel M, Bain G et al (2002) Protection against anoikis and down-regulation of cadherin expression by a regulatable beta-catenin protein. J Biol Chem 277:18677–18686
Whitworth CA, Hudson TE, Rybak LP (1999) The effect of combined administration of cadmium and furosemide on auditory function in the rat. Hear Res 129:61–70
Wong KL, Klaassen CD (1982) Neurotoxic effects of cadmium in young rats. Toxicol Appl Pharmacol 63:330–337
Wu X, Ding D, Sun H, Liu H, Jiang H, Salvi R (2011a) Lead neurotoxicity in rat cochlear organotypic cultures. J Otology 6:45–52
Wu X, Ding D, Sun H, Liu H, Jiang H, Salvi R (2011b) Lead neurotoxicity in rat cochlear organotypic cultures. Chin J Otol 6:44–49
Xu C, Johnson JE, Singh PK, Jones MM, Yan H, Carter CE (1996) In vivo studies of cadmium-induced apoptosis in testicular tissue of the rat and its modulation by a chelating agent. Toxicology 107:1–8
Yang MS, Yu LC, Pat SW (2007) Manipulation of energy and redox states in the C6 glioma cells by buthionine sulfoxamine and N-acetylcysteine and the effect on cell survival to cadmium toxicity. Cell Mol Biol (Noisy-le-grand) 53:56–61
Yu D, Ding D, Jiang H, Stolzberg D, Salvi R (2011) Mefloquine damage vestibular hair cells in organotypic cultures. Neurotox Res 20:51–58
Acknowledgments
Research supported in part by grants from NIH (R01DC009091 and R01DC009219) and NIOSH (R01 OH010235).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liu, H., Ding, D., Sun, H. et al. Cadmium-Induced Ototoxicity in Rat Cochlear Organotypic Cultures. Neurotox Res 26, 179–189 (2014). https://doi.org/10.1007/s12640-014-9461-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-014-9461-4