
Abstract
In the central nervous system, members of the Src family of tyrosine kinases (SFKs) are widely expressed and are abundant in neurons. The purpose of this study is to examine whether glycogen synthase-3 (GSK-3) is involved in SFK inhibitor-induced apoptosis. PP2 and SU6656, SFK inhibitors, increased apoptotic cell death with morphological changes that were characterized by cell shrinkage, chromatin condensation, or nuclear fragmentation. Moreover, both activation of caspase-9 and caspase-3 were accompanied by the cell death. GSK-3 inhibitors, such as alsterpaullone and SB216763, prevented the PP2-induced apoptosis. In addition, insulin-like growth factor-I prevented the PP2-induced cell death and PP2 inhibited phosphorylation of focal adhesion kinase (FAK). Phosphorylation of FAK on Tyr 576 by Src activates FAK. These results suggest that inhibition of SFK induces apoptosis possibly via blocking of FAK/phosphatidylinositol-3 kinase/Akt signaling pathway and activation of GSK-3 is involved in the cell death in rat cortical neurons.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the central nervous system (CNS), members of the Src family of tyrosine kinases (SFKs) are widely expressed, as well as in neurons (Thomas and Brugge 1997). The SFKs are non-receptor protein tyrosine kinases and are involved in neurogenesis, differentiation, migration, neurite extension, and synaptic transmission (Thomas and Brugge 1997; Morgan et al. 2000; Zhao et al. 2003; Kalia et al. 2004).
SFKs have been shown to rescue several cell types from apoptotic death induced by removal of cytokines, irradiation, or chemotherapeutic drugs (Thomas and Brugge 1997; Johnson et al. 2000; Lee et al. 2004). However, it is not clear that SFKs play essential roles in neural cell death.
We have reported that inhibitors of glycogen synthase kinase-3 (GSK-3) reduced caspase-dependent apoptosis in rat cortical neurons, when death was initiated by N-methyl-d-aspartate (NMDA) receptor antagonist (Takadera et al. 2004, 2006). Recent reports suggest that GSK-3 affects many fundamental cellular functions, including the cell cycle, gene transcription, cytoskeletal integrity, and apoptosis (Cross et al. 1995; Grimes and Jope 2001; Hetman et al. 2000). The phosphatidylinositol-3 kinase/Akt signaling pathway is one of the signaling systems implicated in the survival of neurons that leads to inhibition of GSK-3 by increasing Ser9 phosphorylation (Cross et al. 1995; Pap and Cooper 1998). Ca2+-influx through NMDA receptors activates Akt/protein kinase B (PKB) through a PI3-k-dependent pathway and the PI3-k-dependent route to Akt/PKB activation involves possible recruitment of SFK-dependent phosphorylation of a focal adhesion kinase (FAK)–p85 (the regulatory subunit of PI3-k) complex in glutamate stimulation (Crossthwaite et al. 2004).
We, therefore, examined whether SFK inhibitors induce caspase-dependent apoptotic cell death in cultured cells and whether GSK-3 is involved in the SFK inhibitor-induced cell death. We report here that the SFK inhibitors induce apoptosis and that GSK-3 inhibitors block the apoptosis in rat cultured cortical neurons.
Methods
Materials
4-Amino-5-(4-chlorophenyl)-7-(t-butyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2), SU6656 and 4-amino-7-phenylpyrazolo[3,4-d]pyrimidine (PP3) were purchased from Merck (Darmstadt, Germany). Alsterpaullone, protease inhibitor cocktail, and mouse anti-β-actin monoclonal antibody were purchased from Sigma Chemical Co. (St. Louis, USA). Rabbit anti-phospho-GSK-3β (Ser9) antibody, anti-GSK-3β antibody, and anti-FAK antibody were purchased from Cell Signaling Technology, Inc. (Boston, USA). Rabbit (monoclonal) anti-phospho-FAK (Tyr 576) antibody was acquired from GeneTex, Inc. (Irvine, USA). Rabbit anti-phospho-Akt (Ser473) antibody and anti-AKT antibody were purchased from BioSource International, Inc. (Camarillo, USA). Rabbit anti-phospho-Src (Tyr418) was purchased from Invitrogen Co. (Carlsbad, USA). Insulin-like growth factor-I (IGF-I) was acquired from Roche Diagnostics. 3-(2,4-Dichlorophenyl)-4-(1-methyl-1H-indol-3-yl)-1H-pyrrole-2,5-dione (SB216763) was purchased from Tocris Cookson (Bristol, UK). Hoechst 33258 (bis-benzimide) was purchased from Invitrogen (Carlsbad, USA). Ac-Asp-Glu-Val-Asp-4-methyl-coumaryl-7-amide (Ac-DEVD-MCA), Ac-Leu-Glu-His-Asp-4-methyl-coumaryl-7-amide (Ac-LEHD-MCA), and 7-amino-4-methyl-coumarin (AMC) were acquired from the Peptide Institute (Osaka, Japan). Other chemicals were purchased from Wako Pure Industries (Osaka, Japan).
Cell Culture
Cerebral cortical cells were obtained and cultured essentially as described by Dichter (1978) and Choi et al. (1988) from fetal rats (Wistar) after 18–19 days of gestation. Whole cerebral neocortices were removed from fetal mice, taking care to discard the hippocampal formation, basal ganglia, and most of the meninges. The tissue was then minced, incubated in 0.25% trypsin for 30 min at 37°C, and then DMEM plus fetal calf serum was added. The tissue was triturated with Pasteur pipette. Clumps of cells were removed by filtering through a double layer of lens paper.
The dissociated cortical cells were cultured on poly-d-lysine-coated 35 mm dishes (Falcon 3001) (2 × 106 cells/dish) in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal calf serum. 10 μM of cytosine-β-D arabinofuranoside was added to the culture medium on day 3 after plating. The cells were cultured for 9–10 days and then used in the experiments. We examined the relative proportion of neurons to other cell types by staining the cells with anti-neuronal nuclei (NeuN) monoclonal antibody and anti-glial fibrillary protein (GFAP) visualized with an immunohistochemical staining kit, Vectastain ABC kit (Vector). The relative proportion of neurons (NeuN+) to glia cells (GFAP+) was about 95%.
Cell Treatment and Cell Viability
The cells were washed twice with Tris-buffered salt solution containing (in mM): NaCl 120, KCl 5.4, CaCl2 1.8, MgCl2 0.8, Tris–HCl 25, and glucose 15, at pH 6.5, and then replaced with 2 ml of DMEM. We used washing buffer at pH 6.5 to block the glutamate neurotoxicity during the treatment. Reagents such as PP2 were then added to the cells. The treatment was carried out for 1–2 days at 37°C. Morphological cell changes were observed by phase-contrast microscopy during the experiment.
Apoptotic cell death was determined by staining the cells with Hoechst dye H33258. Cells were fixed with a 10% formalin neutral phosphate buffer solution (pH 7.4) for 5 min at room temperature. After washing the cells with distilled water, they were stained with 8 μg/ml H33258 for 5 min. The nuclear morphology was observed under a fluorescent microscope (Olympus IX70 model). Apoptosis was quantitated by scoring the percentage of cells with apoptotic nuclear morphology at the single cell level. Condensed or fragmented nuclei were scored as apoptotic. A total of 5–7 randomly selected fields were captured using Polaroid PDMC II software. At least 200 cells were counted per condition, and each experiment was repeated three times.
Caspase Activity
The caspase activity was measured as described previously (Takadera and Ohyashiki 1997). In brief, the cells were washed with phosphate-buffered saline and suspended in 50 mM Tris–HCl buffer (pH 7.4) containing 1 mM EDTA and 10 mM EGTA. The cells were treated with 10 μM digitonin for 10 min. The lysates were obtained by centrifugation at 10,000×g for 5 min, and the cleared lysates containing 50–100 μg of protein were incubated with 50 μM enzyme substrate Ac-LEHD-MCA (caspase-9) and Ac-DEVD-MCA (caspase-3) for 1 h at 37°C. The specificities of the peptide substrates have been reported (Thornberry et al. 1997). The reaction was terminated by addition of monoiodoacetic acid (5 mM). AMC levels were measured using a spectrofluorometer (Hitachi F4500, Japan) with excitation at 380 nm and emission at 460 nm, and the activity is expressed as pmol or nmol of AMC released/min/mg of protein.
Western Blotting
Primary cultured cells were scraped off the dish and collected by centrifugation (400×g for 5 min) followed by homogenization in ice-cold buffer (50 mM Tris–HCl buffer containing 50 mM NaCl, 10 mM EGTA, 5 mM EDTA, 2 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 1 mM benzamide, and 10% protease inhibitor cocktail, pH 7.4). The cell suspension was placed on ice for 30 min and centrifuged at 18,000×g for 30 min. The supernatant was diluted with an equal volume of sample buffer containing 62.5 mM Tris–HCl (pH 6.8), 2% sodium dodecyl sulfate (SDS), 10% glycerol, 50 mM dithiothreitol, and 0.1% bromophenol blue, heated at 95°C for 5 min, and stored at −20°C. Protein concentration was determined by the bicinchoninic acid assay. Each sample (20–30 μg/lane) was loaded and separated using 10% SDS-polyacrylamide gel electrophoresis. Proteins were transferred on a nitrocellulose membrane and blocked with Tris-buffered saline containing 5% skim milk for 1 h at room temperature and then incubated with anti-Akt, anti-phospho-Akt, anti-GSK-3β, anti-phospho-GSK-3β, anti-phospho-Src, or anti-phospho-FAK antibody in Tris-buffered saline overnight at 4°C. After washing for 5 min with three changes of Tris-buffered saline, the membrane was incubated with a phosphatase-conjugated goat anti-rabbit or mouse antibody for 1 h at room temperature in Tris-buffered saline. After washing for 5 min with three changes of Tris-buffered saline, immunoreactive bands were visualized with a Western blot detection kit BCIP/NBT system. Equal amounts of protein extracts were also analyzed by Western blot analysis with anti-actin antibody.
RT-PCR
RT-PCR was carried out using the Access RT-PCR System (Promega, Madison, USA). Total RNA was isolated from cortical cells on day 10 by the guanidine isothiocyanate method using an RNeasy mini kit (QIAGEN GmbH, Germany) according to the manufacturer’s directions. The reverse transcription reaction was performed at 48°C for 45 min. After incubation at 95°C for 9 min, 30 cycles of amplification (30 s at 94°C (denaturation), 1 min at 60°C (annealing), and 2 min at 68°C (extension)) were performed using the following oligonucleotides: 5′-GGTATTGAGACAGACAGTGG-3′ (caspase-3 sense primer) and 5′-CATGGGATCTGTTTCTTTGC-3′ (caspase-3 antisense primer).
Statistical Analysis
Statistical significance was assessed by one-way ANOVA and post-hoc Scheffe’s comparisons.
Results
Apoptosis Induced by SFK Inhibitors
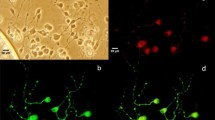
We examined the effect of PP2, a selective SFK inhibitor, on rat cultured cortical cells. Cell treatment with 5 μM PP2 induced apoptotic morphology, such as shrunken cell bodies, fragmented processes, and condensed or fragmented nuclei (Fig. 1). Cell viability was checked by staining with both trypan blue and Hoechst 33258. More than 50% of cells showed apoptotic cell death by treatment with 5 μM PP2 for 48 h (Fig. 2a). PP3, a negative control for the Src family of protein kinase inhibitor PP2, did not induce cell death (Fig. 2a). Dimethyl sulfoxide (DMSO), in which PP2 was dissolved, had no effect on cell viability and nuclear morphology in cortical cells (data not shown). Using an antibody that recognizes activated Src, which is phosphorylated on Tyr418 in the catalytic domain (Cooper and MacAuley 1988), we assessed that PP2 (5 μM) blocked Src activity in the cortical cells (Fig. 2b). Another SFK inhibitor, SU6656, also induced apoptotic cell death in a dose-dependent manner (Fig. 2c).

SFK inhibitor-induced apoptosis. Phase-contrast (a, b) and H33258 fluorescence (c, d) microscopy of the cortical cells after exposure to the SFK inhibitor, PP2. Cells were incubated with 0.1% DMSO (a, c) or 5 μM PP2 (b, d) for 48 h at 37°C as described in “Methods” section. Scale bars = 10 μm. Arrowheads indicate healthy neurons. Arrows indicate neurons with apoptotic morphology (condensed or fragmented nuclei)

a Time-dependent SFK inhibitor-induced cell death. The cells were incubated with or without PP2 (5 μM) or PP3 (5 μM) for 12–48 h at 37°C. Apoptotic cell death was checked by staining with Hoechst 33258 as described in “Methods” section. Apoptosis was quantitated by scoring the percentage of cells with apoptotic nuclear morphology at the single cell level. Condensed or fragmented nuclei were scored as apoptotic. Data are shown as the mean ± SEM. n = 3 per group. *P < 0.05 (vs. control cells). b Effect of PP2 on Src phosphorylation levels. The cells were incubated in the presence or absence of PP2 (5 μM) for 1 h at 37°C. Cells were then lyzed and the lysate was immunoblotted with anti-Src (Tyr418) or anti-actin antibody. Data are expressed as a percentage of optical density value for control. Data are shown as the mean ± SEM. n = 3 per group. *P < 0.05 (vs. control cells). c SFK inhibitor (SU6656)-induced apoptosis. The cells were incubated with SU6656 (1–10 μM) in the presence or absence of for 48 h at 37°C. Apoptotic cell death was measured as described in “a”. Each value represents the mean ± SEM. *P < 0.05 (vs. control cells)
Caspases are known to be involved in apoptosis in many injured cells. Knock-out mice have been used to highlight the critical importance of caspase-9, an initiator caspase, and caspase-3, an effector caspase, in neuronal apoptosis during development (Kuida et al. 1996, 1998; Hakem et al. 1998). Therefore, we measured caspase-9 and caspase-3 activity after treatment of the cells with PP2 and PP3 using peptide substrates. Treatment of the cells with 5 μM PP2 for 12 h increased the activities of caspase-9 and caspase-3 (Fig. 3a, b). The negative control PP3 had no effect on the activities of caspase-3 and caspase-9. The expression of caspase-3 was detected in these cells using RT-PCR methods in the rat cortical cells (Fig. 3c).

a PP2-induced caspase-9 activation. b PP2-induced caspase-3 activation. The cells were incubated with or without 5 μM PP2 or PP3 for 12 h at 37°C. Data are shown as the mean ± SEM. n = 3 per group. *P < 0.05 (vs. control cells). Caspase activity was measured as described in “Methods” section using peptide substrates. Caspase-9 activity and caspase-3 activity were measured using Ac-LEHD-MCA and Ac-DEVD-MCA, respectively. c Detection of caspase-3 mRNA by RT-PCR. Amplified products were electrophoresed in 2% agarose gel. The expected product for caspase-3 is 281 bp long (lane 2). Lanes 1 and 3, size marker of 100-bp DNA ladder. Lane 4, no reverse transcriptase reaction
The apoptotic cell death induced by 5 μM PP2 was attenuated by the presence of 0.1 μg/ml cycloheximide, indicating that the synthesis of new proteins is required for PP2-induced apoptosis, a characteristic of programmed cell death (Fig. 4).

Effect of cycloheximide on PP2-induced apoptosis. The cells were incubated with PP2 (5 μM) in the presence or absence of cycloheximide (0.1 μg/ml) for 48 h at 37°C. Apoptotic cell death was measured as described in Fig. 2a. Each value represents the mean ± SEM. *P < 0.05 (vs. PP2-only-treated cells)
Insulin Growth Factor-I (IGF-I) Protected the Cells from PP2-Induced Cell Death
IGF-I has been reported to play a role in differentiation and survival in CNS via the PI3-K/Akt signaling pathway (Stewart and Rotwein 1996). We reported previously that IGF-I attenuated NMDA receptor antagonist-induced apoptotic cell death in the rat cortical cells (Takadera et al. 1999). As shown in Fig. 5, IGF-I at 50 ng/ml completely inhibited PP2-induced apoptosis.

Effect of IGF-I on PP2-induced apoptosis. The cells were incubated with PP2 (5 μM) in the presence or absence of IGF-I (50 ng/ml) for 48 h at 37°C. Apoptotic cell death was measured as described in Fig. 2a. Each value represents the mean ± SEM. *P < 0.05 (vs. PP2-only-treated cells)
PP2 Decreased Akt Phosphorylation Levels
Akt is phosphorylated at two sites that are associated with activation of enzyme activity: Thr308 in the catalytic domain and Ser473 in the cytoplasmic domain. Phosphorylation of both sites is critically dependent upon PI3-K activity (Alessi et al. 1997). Akt phosphorylation at Ser473 was determined by immunoblot analysis utilizing a phospho-Akt-(Ser473)-specific antibody. PP2 (5 μM) reduced phospho-Akt levels (Fig. 6).

Effect of PP2 on Akt phosphorylation levels. The cells were incubated in the presence or absence of PP2 (5 μM) for 1 h at 37°C. Cells were then lyzed and the lysate was immunoblotted with anti-Akt or anti-phospho-Akt (Ser473) antibody. Data are expressed as a percentage of optical density value for control. Data are shown as the mean ± SEM. n = 3 per group. *P < 0.05 (vs. control cells)
GSK-3 Inhibitors Prevented PP2-Induced Apoptosis
GSK-3 is a principal physiological substrate of Akt and the activity of GSK-3 is inhibited by Akt-mediated phosphorylation in response to trophic stimulation such as IGF-I. To investigate directly the role of the activity of endogenous GSK-3 in the cell death in response to the SFK inhibitor treatment, we assayed the effect of selective inhibitors, alsterpaullone and SB216763, on PP2-induced apoptosis. The GSK-3 inhibitors showed a protective effect against the PP2-induced apoptosis (Fig. 7a, b).

a Effect of SB216763 on PP2-induced apoptosis. The cells were incubated with PP2 (5 μM) in the presence or absence of SB216763 (0.5–5 μM) for 48 h at 37°C. Each value represents the mean ± SEM. *P < 0.05 (vs. PP2-only-treated cells). Apoptotic cell death was measured as described in Fig. 2a. b Effect of alsterpaullone on PP2-induced apoptosis. The cells were incubated with PP2 (5 μM) in the presence or absence of alsterpaullone (0.5–2 μM) for 48 h at 37°C. Apoptotic cell death was measured as described in Fig. 2a. Each value represents the mean ± SEM. *P < 0.05 (vs. PP2-only-treated cells). Als alsterpaullone
The kinase activity of GSK-3β is inhibited by phosphorylation on serine-9. We then examined the effect of PP2 on the phosphorylation of GSK-3β on serine-9 using a specific antibody detecting the phosphorylation of GSK-3β on serine-9. As shown in Fig. 8, GSK-3β dephosphorylation was detected in PP2-treated cells.

Effect of PP2 on GSK-3 phosphorylation levels. The cells were incubated in the presence or absence of PP2 (5 μM) for 1 h at 37°C. Cells were then lyzed and the lysate was immunoblotted with anti-GSK-3β or anti-phospho-GSK-3β (Ser9) antibody. Data are expressed as a percentage of optical density value for control. Data are shown as the mean ± SEM. n = 3 per group. *P < 0.05 (vs. control cells)
PP2 Decreased FAK Phosphorylation Levels
Phosphorylation of FAK on Tyr 576 by Src activates FAK (Calalb et al. 1995). The PI3-K p85 subunit interacts with FAK via its SH2 domain and is activated by FAK (Girault et al. 1999; Xia et al. 2004). We, therefore, examined the effect of PP2 on the phosphorylation of FAK on Tyr576 in the rat cortical cells. As shown in Fig. 9, PP2 markedly decreased the FAK phosphorylation levels.

Effect of PP2 on FAK phosphorylation levels. The cells were incubated in the presence or absence of PP2 (5 μM) for 1 h at 37°C. Cells were then lyzed and the lysate was immunoblotted with anti-FAK or anti-phospho-FAK (Tyr576) antibody. Data are expressed as a percentage of optical density value for control. Data are shown as the mean ± SEM. n = 3 per group. *P < 0.05 (vs. control cells)
Discussion
We showed in this report that the GSK-3 inhibitors protected cortical neurons from SFK inhibitor-induced apoptosis, suggesting that GSK-3 activity is critical for neuronal cell death induced by inhibiting Src-family tyrosine kinase activity.
We previously reported that cycloheximide protects apoptosis induced by blocking the trophic effect of NMDA receptor in rat cortical cells (Takadera et al. 1999). Similarly, cycloheximide protected PP2-induced apoptosis, suggesting that PP2-induced apoptosis requires synthesis of proapoptotic protein(s) downstream of the apoptotic pathway (Fig. 4).
The mechanism by which Src inhibitors induce apoptosis in the rat cortical cells is not clear. NMDA receptor antagonist triggers rat or mouse cortical neuron apoptosis in immature rodent, suggesting that NMDA receptor has pro-survival activity during CNS development (Takadera et al. 1999, 2004; Ikonomidou et al. 1999; Hwang et al. 1999). Non-receptor tyrosine kinases of the Src family are closely associated with NMDA receptor (Wang and Salter 1994). Ca2+-influx through NMDA receptors activates Akt/protein kinase B (PKB) through a phosphatidylinositol 3-kinase (PI 3-kinase)-dependent pathway and the PI 3-kinase-dependent route to Akt/PKB activation involves possible recruitment of an SFK-dependent phosphorylation of a FAK–p85 (the regulatory subunit of PI 3-kinase) complex in glutamate stimulation (Crossthwaite et al. 2004).
FAK has an important role in the prevention of apoptosis by cell attachment. FAK could have a similar function in the nervous system (Girault et al. 1999). Phosphorylation of FAK on Tyr 576 by Src activates FAK (Calalb et al. 1995). The PI3-K p85 subunit interacts with FAK via its SH2 domain and might be activated by FAK (Girault et al. 1999). As shown in Fig. 9, PP2 inhibited the phosphorylation of FAK on Tyr576. The SFK inhibitor may interfere with the survival signal of NMDA receptor via inhibition of FAK/PI3-K/Akt signaling pathway and induce GSK-3-dependent apoptosis.
Glycogen synthase kinase-3 activity is known to be suppressed when it becomes phosphorylated on serine 9 by activation of Akt (Cross et al. 1995; Pap and Cooper 1998). IGF-I has been reported to play a role in differentiation and survival in CNS (Stewart and Rotwein 1996) and to activate the phosphatidylinositol-3 kinase (PI3-K)/Akt signaling pathway (Alessi et al. 1997). As expected, IGF-I completely inhibited PP2-induced apoptosis (Fig. 5).
The critical importance of caspase-9, an initiator caspase, and caspase-3, an effector caspase, in neuronal apoptosis during development has been shown using knock-out mice (Kuida et al. 1996, 1998; Hakem et al. 1998). The apoptosis induced by the SFK inhibitor was accompanied by activation of caspase-9 and caspase-3. We have reported that NMDA antagonist-induced caspase-3 activation is blocked by GSK-3 inhibitors, suggesting that GSK-3 probably acts at a site upstream of caspase-3 (Takadera et al. 2006).
However, the downstream substrates of GSK-3 that ultimately induce neuronal death are not clear. Linseman et al. (2004) have reported that GSK-3 phosphorylates Bax, a pro-apoptotic Bcl-2 family member, and promotes its mitochondrial localization. Bax stimulates the intrinsic (mitochondrial) death pathway, including caspase cascade, by eliciting cytochrome c release from mitochondria,
We showed in this report for the first time that GSK-3 inhibitors protected cortical neurons from SFK inhibitor-induced apoptosis.
References
Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P (1997) Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol 7:261–269
Calalb MB, Polte TR, Hanks SK (1995) Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol 15:954–963
Choi DW, Koh JY, Peters S (1988) Pharmacology of glutamate neurotoxicity in cortical cell culture: attenuation by NMDA antagonists. J Neurosci 8:185–196
Cooper JA, MacAuley A (1988) Potential positive and negative autoregulation of p60c-src by intermolecular autophosphorylation. Proc Natl Acad Sci USA 85:4232–4236
Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378:785–789
Crossthwaite AJ, Valli H, Williams RJ (2004) Inhibiting Src family tyrosine kinase activity blocks glutamate signalling to ERK1/2 and Akt/PKB but not JNK in cultured striatal neurones. J Neurochem 88:1127–1139
Dichter MA (1978) Rat cortical neurons in cell culture: culture methods, cell morphology, electrophysiology, and synapse formation. Brain Res 149:279–293
Girault JA, Costa A, Derkinderen P, Studler JM, Toutant M (1999) FAK and PYK2/CAKbeta in the nervous system: a link between neuronal activity, plasticity and survival? Trends Neurosci 22:257–263
Grimes CA, Jope RS (2001) The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol 65:391–426
Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, Elia A, de la Pompa JL, Kagi D, Khoo W, Potter J, Yoshida R, Kaufman SA, Lowe SW, Penninger JM, Mak TW (1998) Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell 94:339–352
Hetman M, Cavanaugh JE, Kimelman D, Xia Z (2000) Role of glycogen synthase kinase-3beta in neuronal apoptosis induced by trophic withdrawal. J Neurosci 20:2567–2574
Hwang JY, Kim YH, Ahn YH, Wie MB, Koh JY (1999) N-Methyl-d-aspartate receptor blockade induces neuronal apoptosis in cortical culture. Exp Neurol 159:124–130
Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vockler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW (1999) Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 283:70–74
Johnson D, Agochiya M, Samejima K, Earnshaw W, Frame M, Wyke J (2000) Regulation of both apoptosis and cell survival by the v-Src oncoprotein. Cell Death Differ 7:685–696
Kalia LV, Gingrich JR, Salter MW (2004) Src in synaptic transmission and plasticity. Oncogene 23:8007–8016
Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA (1996) Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature 384:368–372
Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA (1998) Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 94:325–337
Lee M, Kim JY, Koh WS (2004) Apoptotic effect of PP2 a Src tyrosine kinase inhibitor, in murine B cell leukemia. J Cell Biochem 93:629–638
Linseman DA, Butts BD, Precht TA, Phelps RA, Le SS, Laessig TA, Bouchard RJ, Florez-McClure ML, Heidenreich KA (2004) Glycogen synthase kinase-3beta phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J Neurosci 24:9993–10002
Morgan JC, Majors JE, Galileo DS (2000) Wild-type and mutant forms of v-src differentially alter neuronal migration and differentiation in vivo. J Neurosci Res 59:226–237
Pap M, Cooper GM (1998) Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-kinase/Akt cell survival pathway. J Biol Chem 273:19929–19932
Stewart CE, Rotwein P (1996) Growth, differentiation, and survival: multiple physiological functions for insulin-like growth factors. Physiol Rev 76:1005–1026
Takadera T, Ohyashiki T (1997) Apoptotic cell death and caspase 3 (CPP32) activation induced by calcium ionophore at low concentrations and their prevention by nerve growth factor in PC12 cells. Eur J Biochem 249:8–12
Takadera T, Matsuda I, Ohyashiki T (1999) Apoptotic cell death and caspase-3 activation induced by N-methyl-d-aspartate receptor antagonists and their prevention by insulin-like growth factor I. J Neurochem 73:548–556
Takadera T, Sakamoto Y, Ohyashiki T (2004) NMDA receptor 2B-selective antagonist ifenprodil-induced apoptosis was prevented by glycogen synthase kinase-3 inhibitors in cultured rat cortical neurons. Brain Res 1020:196–203
Takadera T, Ishida A, Ohyashiki T (2006) Ketamine-induced apoptosis in cultured rat cortical neurons. Toxicol Appl Pharmacol 210:100–107
Thomas SM, Brugge JS (1997) Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol 13:513–609
Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, Chapman KT, Nicholson DW (1997) A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J Biol Chem 272:17907–17911
Wang YT, Salter MW (1994) Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature 369:233–235
Xia H, Nho RS, Kahm J, Kleidon J, Henke CA (2004) Focal adhesion kinase is upstream of phosphatidylinositol 3-kinase/Akt in regulating fibroblast survival in response to contraction of type I collagen matrices via a beta 1 integrin viability signaling pathway. J Biol Chem 279:33024–33034
Zhao WQ, Alkon DL, Ma W (2003) c-Src protein tyrosine kinase activity is required for muscarinic receptor-mediated DNA synthesis and neurogenesis via ERK1/2 and c-AMP-responsive element-binding protein signaling in neural precursor cells. J Neurosci Res 72:334–342
Acknowledgment
This study was supported in part by a special in-house research grant from Hokuriku University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Takadera, T., Fujibayashi, M., Koriyama, Y. et al. Apoptosis Induced by Src-Family Tyrosine Kinase Inhibitors in Cultured Rat Cortical Cells. Neurotox Res 21, 309–316 (2012). https://doi.org/10.1007/s12640-011-9284-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-011-9284-5