
Abstract
Abuse of amphetamine analogues, such as methamphetamine (METH), represents an important health problem because of their powerful addictive and neurotoxic effects. Abuse of METH induces dopamine neuron terminals loss and cell death in the striatum similar to what is found in other neurodegenerative processes. Exposing mice and rats to enriched environments (EE) has been shown to produce significant protective effects against drug-induced reward as well as against neurodegenerative processes. Here, we investigated whether exposure to EE could reduce the METH-induced reward and neurotoxicity. For this, we reared mice for 2 months during early stages of life in standard environments or EE and then, at adulthood, we tested the ability of METH to induce conditioned place preference and neurotoxicity. We found that, contrary to what we found with other drugs such as cocaine and heroin, EE was unable to reduce the rewarding effects of METH. In addition, contrary to what we found with other toxins such as MPTP, EE did not diminish the striatal neurotoxicity induced by METH (4 × 10 mg/kg) as measured by dopamine content, tyrosine hydroxylase protein levels and apoptosis. Our results demonstrate that the rewarding and neurotoxic effects of METH are not reduced by EE and highlight the great risks associated with the increased popularity of this drug amongst the young population.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The illicit drug methamphetamine (METH) is a substituted amphetamine whose abuse has increased worldwide at an alarming rate over the last decade (Krasnova and Cadet 2009). The chronic use of METH is often associated with cognitive deficits, impaired judgement, aggressive behaviour, anxiety and/or psychosis (Nordahl et al. 2003; Scott et al. 2007, for review). These neuropsychiatric complications might be, at least in part, related to drug-induced neuropathological changes in the brain of METH-exposed individuals (Scott et al. 2007). Human brain imaging studies have provided evidence for significant changes in brain dopaminergic and serotonergic neurotransmitter systems, as well as altered general metabolic activity in basal ganglia and frontal cortices (Chang et al. 2007, for review). Chronic METH use induces significant decreases in dopamine, dopamine transporter (DAT) and dopamine synthesizing enzyme, tyrosine hydroxylase (TH) levels in the caudate and putamen (Wilson et al. 1996; Aron and Paulus 2007). In animals, multiple high-dose (‘binge’) METH regimens have been used to model the METH-induced neurotoxicity because they produce extensive damage of dopamine and serotonin terminals with reductions in striatal dopamine content, DAT density, and TH activity (Davidson et al. 2001; Cadet et al. 2003; Volz et al. 2007, for review). In addition to these neurotoxic effects on terminals, others and we have recently shown that METH induces cell death of striatal GABA neurons in mice through apoptotic processes (Deng and Cadet 2000; Thiriet et al. 2005).
Vulnerability to drugs depends on complex interactions between the drug, genetic and environmental factors. Enriched environments (EE), a combination of both inanimate and social stimulations (Rosenzweig and Bennett 1996), appear to have beneficial effects in several models of addiction and neurotoxicity. Exposure to EE has been shown to have biochemical and structural consequences in several brain regions particularly in the cortex, the hippocampus and the striatum (Diamond et al. 1976; Kempermann et al. 1997; van Praag et al. 2000; Nithianantharajah and Hannan 2006). These alterations are due, at least in part, to changes in gene expression produced by environmental stimulation (Rampon et al. 2000; Bezard et al. 2003; Thiriet et al. 2008; Solinas et al. 2009). In relation with this study, we have shown that mice reared in EE have lower striatal levels of DAT (Bezard et al. 2003), the molecular target of METH (Barr et al. 2006) and accumulating evidences demonstrate that EE reduce the vulnerability to drugs (Laviola et al. 2008; Carroll et al. 2009; Stairs and Bardo 2009). For example, using a conditioned place preference (CPP) paradigm, we observed that the rewarding effects of cocaine and heroin are blunted in mice reared in EE compared to mice raised in standard environments (SE) (El Rawas et al. 2009; Solinas et al. 2009).
Interestingly, vulnerability or resistance to neuronal toxins also seems to depend on environmental factors. For instance, unpredictable or chronic stressful conditions increase the neurotoxicity of METH (Matuszewich and Yamamoto 2004; Tata et al. 2007). In contrast, positive life experiences appear to have beneficial effects in models of several brain injuries, such as ischaemia, epilepsy or neurodegenerative diseases (van Praag et al. 2000; Nithianantharajah and Hannan 2006; Laviola et al. 2008). Indeed, in a transgenic mouse model of Huntington’s disease, EE delay the onset of progression of motor symptoms as well as the cognitive deficits (Nithianantharajah et al. 2008). In addition, in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) model of PD, mice reared in EE are resistant to MPTP’s toxic effects on dopamine terminals (Bezard et al. 2003).
We undertook this study to investigate the potential beneficial effect of EE against the METH-induced reward and toxicity in mice. We analysed the rewarding effects of METH using a paradigm of place preference conditioning and its neurotoxicity by analysing both METH-induced reductions in the striatal levels of dopamine and of TH, a key enzyme in dopamine synthesis, and METH-induced cell death in the striatum. We focussed on this brain region because it appears to be central for the effects of both METH (Fleckenstein et al. 2000; Cadet et al. 2003; Riddle et al. 2005) and EE (Bezard et al. 2003; Laviola et al. 2008; Thiriet et al. 2008; Solinas et al. 2009).
Materials and Methods
Subjects
Male C57Bl/6J mice were housed in a temperature-controlled environment on a 12-h light/12-h dark cycle with the lights on from 7:00 a.m. to 7:00 p.m. and had ad libitum access to food and water. All experimentation was conducted during the light period. Experiments were carried out in accordance with the European Communities Council Directive of 24 November 1986 (86/609/EEC) for the care of laboratory animals.
Housing Conditions
Two distinct housing environmental conditions were used for this study, SE or EE. SE consisted of common cage housing (30 × 20 × 15 cm) whilst EE consisted of larger (60 × 38 × 20 cm) cages containing constantly a running wheel, a small house and four-five toys that were changed once a week with new toys of different shapes and colours. For both SE and EE, mice were housed in groups of 4. Mice were placed in SE or EE immediately after weaning and for 2 months to expose them to these environments during the entire duration of their adolescence. They were then subjected to CPP paradigm or multiple high-dose (‘binge’) METH regimens. This protocol of enrichment was previously used in our group and has revealed beneficial impact both against rewarding properties of another psychostimulant, namely cocaine, or another class of drug heroin (Solinas et al. 2008; El Rawas et al. 2009).
Conditioned Place Preference
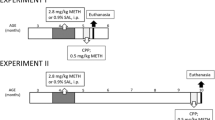
CPP was performed as previously described (Solinas et al. 2008; El Rawas et al. 2009; Solinas et al. 2009). Four identical boxes formed by two lateral chambers (15 × 15 × 20 cm) connected by a central alley (5 × 15 × 20 cm) were used (IMETRONIC, Pessac, France). Two sliding doors separated the alley from the chambers. In each chamber, two different metallic grids, one with large (1 cm) squares and the other with small (0.5 cm) circles were used as conditioned stimuli. Two infrared photocells were present in each compartment and detected the presence and movements of mice. CPP procedure consisted of three phases: preconditioning, conditioning and test. For each manipulation, mice were brought to the experimental room 60 min before the start of the experiment to allow for habituation and to reduce stress. Preconditioning was performed on day 1. Mice were placed in the central compartment with the doors closed. After 15 s, the doors were opened and mice were free to explore the entire apparatus for 30 min. The time spent in each compartment was recorded and was considered a measure of spontaneous preference. Conditioning was performed on days 2, 3 and 4. Conditioning sessions were performed twice per day with morning and afternoon sessions separated by at least 6 h. During these sessions, mice were injected with either saline in the morning and d-methamphetamine-HCl (METH) (0.5 or 1 mg/kg) in the afternoon and immediately confined to one of the pairing compartments for 30 min. METH was kindly provided by the National Institute on Drug Abuse (USA). The compartments (right or left) were counterbalanced. Control mice received saline on both sides of the cage. Test for CPP was performed on day 5. These test sessions were conducted similarly to preconditioning sessions with animals placed in the central compartment for 15 s and then left free to choose a compartment for 30 min. The time spent in each compartment was measured and compared to the time spent in the same compartment during the preconditioning session. Preference scores in seconds were calculated by subtracting the time spent during the preconditioning day from the time spent during the test day in the compartment paired to METH injection and constituted a measure of the preference developed for this compartment and of the rewarding effects of METH. Locomotor activity during conditioning sessions was measured by the total number of interruptions of photocell beams (locomotor counts). For the saline control group, we verified that the data for CPP and locomotor activity were similar in the right and left compartments and then arbitrarily chose to use data from the left compartment for graphic representation and statistical analysis.
Multiple high-dose (‘binge’) METH treatment
At adulthood (about 3 months of age), mice received four intra-peritoneal injections of METH or saline every 2 h. To evaluate the neurotoxicity of METH, and in line with our previous studies on METH toxicity, we injected mice with METH at a dosage (4 × 10 mg/kg, at 2 h intervals) that produces significant loss of striatal dopamine and cell death (Davidson et al. 2001; Thiriet et al. 2005). In addition, in this study, we also analysed the effect of a lower dose of METH (4 × 5 mg/kg, at 2 h intervals) on striatal dopamine and DOPAC content to rule out the possibility that positive effects of EE could be found with this lower, but toxic (Layer et al. 1993), dosage of METH. All drugs were injected in a volume of 1 ml/100 g.
Body Temperature Measurement
Because METH-induced changes in body temperature are believed to participate in its toxicity (Cadet et al. 2003), we monitored rectal temperature in a different set of mice that did not undergo biochemical analysis. On the day of experiment, animals were placed in experimental room for 1 h before any manipulation. A rectal probe digital thermometer (BIO-9882, Bioseb, France) was inserted into the rectum of each mouse where it remained for approximately 5 s until stable temperature was obtained. Rectal measurements were recorded before the first injection of METH or saline and then 1 h after each injection of METH (10 mg/kg) or saline (time of measurements = 0, 1, 3, 5, 7 h).
Striatal Dopamine and DOPAC Content
The animals were sacrificed 21 days after drug treatment by decapitation because dopamine loss is at its maximum at this time-point (O’Callaghan and Miller 1994). Brains were quickly removed and striata were dissected over ice. Striatal tissues were then immediately frozen on dry ice and stored at −80°C until HPLC (high performance liquid chromatography) analysis. One hemi-striatum per animal was used for tissue level analysis of dopamine and its metabolite 3,4-dihydroxyphenylacetic acid (DOPAC). For this, brain tissues were weighed and homogenized by sonication on ice in 0.1 M perchloric acid (10 μl/mg of tissue), containing 10−6 M of dihydroxybenzoic acid, as an internal standard, and centrifuged at 13,000 rpm for 10 min at 4°C. After filtration through 0.22-μm filters (Millipore, France), 10 μl of samples was injected in a HPLC column (Teknokroma Kromasil 100 C18, 5 μm, 0.4 × 250 mm) coupled to an electrochemical detector (Esa Coulochem III). The mobile phase used was prepared with NaH2PO4 100 mM, methanol 25%, EDTA 0.2 mM and octyl sulphate 0.7 mM, pH 3. To determine dopamine and DOPAC tissue levels, we compared the heights of the peaks in mice samples to those of standard solution containing dopamine and DOPAC. Monoamine levels were calculated as ng/mg of tissue weight.
Anti-TH Western Blotting
Animals were sacrificed 21 days after drug treatment by decapitation; their brains were quickly removed, brain regions were dissected over ice, immediately frozen on dry ice and stored at −80°C until processed for western blot experiments. One hemi-striatum per animal was homogenized in a buffer (350 μl/25 mg) containing Tris HCl 0.2 M, pH 6.9, SDS 8% and glycerol 35% and heated to 90°C for 3 min. Samples were then centrifuged (13,000 rpm, 10 min) and supernatants were analysed for protein content using DC Protein assay (BioRad, France). 50 μg of protein per sample were loaded on a SDS page gel. Gels were transferred to polyvinylidene difluoride membranes. Membranes were blocked in Tris-buffered saline (TBS), containing 3% Bovine serum albumin (BSA) and 0.1% Tween 20 and then incubated with two different antibodies: (1) mouse anti-tyrosine hydroxylase (TH) antibody (1/5,000) (Euromedex, France) and (2) rabbit anti-β actin (1/10,000) (Sigma, France) diluted in the same buffer for 2 h at room temperature (RT). Blots were then rinsed 3 × 10 min in TBS containing 0.1% of Tween 20. Incubation with second antibodies goat anti-mouse coupled to Alexa Fluor 568 and goat anti-rabbit coupled to Alexa Fluor 488 (1/1,000, Molecular Probes) diluted in TBS containing 3% BSA and 0.1% Tween 20 took place for 1 h at RT. Membranes were rinsed as described above and analysed using Typhoon Imaging system (GE Healthcare). After quantification using image J software, TH signal was normalized to β-actin signal to avoid any loading artefact.
Terminal Deoxynucleotidyl Transferase-Mediated Deoxyribonucleotide Triphosphate Nick End Labelling
The animals were sacrificed 3 days after drug treatment by decapitation, a time point known to produce a maximal apoptotic signal (Deng et al. 1999). The brains were quickly removed, immediately frozen on dry ice, and stored at −80°C until used. Slide-mounted sections (25 μm) were fixed by incubation in paraformaldehyde 4% (4°C, 30 min) and rinsed in water 3 × 5 min at RT. To increase permeability, sections were incubated in 0.5% Triton X-100 diluted in PBS, pH 7.4, 20 min at 80°C and rinsed 3 × 5 min in PBS at RT. Slices were then incubated 30 min at RT in PBS containing 0.3% H2O2 and rinsed 3 × 5 min in PBS at RT. To label apoptotic nuclei, sections were incubated with 50 μl of fluorescein labelled dUTP (Roche, France) containing 0.5 μl terminal transferase (TdT) (New England Biolabs, France) for 1 h at 37°C and rinsed 3 × 5 min in PBS at RT. Negative (without TdT) and positive (DNAse treatment) controls were performed to validate signals. To label all nuclei, sections were exposed for 5 min to propidium iodide (0.1 μg/ml) diluted in PBS, rinsed 3 × 5 min in PBS at RT and coverslipped by using Mowiol mounting solution. Using fluorescent microscope, fluorescein positively labelled cells from four striatum per mouse (right and left sides of 2 sections) taken at bregma = 1.00 ± 0.30 were counted by an individual blind to the experimental treatment (6–10 animals were included per group) as previously done (Thiriet et al. 2005). Representative pictures were taken using a confocal laser microscope (BX60, Olympus).
Statistical Analysis
Data are presented as mean ± SEM. Statistical analysis was performed using two-way analysis of variance (ANOVA) (StatView 4.02, SAS Institute, Cary, NC) with environment (EE and SE) and treatment (METH and saline) as factors. Results showing significant overall changes were subjected to Student–Newman–Keuls post hoc test. The null hypothesis was rejected at P < 0.05.
Results
Exposure to EE does not Reduce the Activating and Rewarding Effects of METH
During pre-tests, both SE and EE mice showed no significant natural preferences for either CPP compartments (data not shown). During conditioning sessions, in the CPP apparatus, we measured locomotor activity after injections of saline or METH in saline controls mice (Fig. 1a), in mice that were conditioned to METH 0.5 mg/kg (Fig. 1b) and in mice that were conditioned to METH 1.0 mg/kg (Fig. 1c). In all conditioning groups, locomotor activity after saline injections decreased over conditioning sessions (Fig. 1a–c). Consistent with previous studies (Bezard et al. 2003), EE mice showed reduced locomotor activity after injections of saline compared to SE mice (Fig. 1a–c). Locomotor activity after injections of METH 0.5 mg/kg remained stable across sessions and became significantly different from the activity measured after saline injections on the third day of conditioning (Fig. 1b). Injections of METH (1.0 mg/kg) induced high levels of locomotor activity that remained stable across conditioning sessions (Fig. 1c). No evidence for sensitization of the activating effects of METH was found at either doses of METH. Importantly, the effects of METH on locomotor activity did not differ between SE and EE at any dose tested. For saline injections, statistical analysis revealed a significant effect of the environment (F 1,20 = 15.21, P < 0.001) and of day (F 2,40 = 12.92, P < 0.0001) but no environment × day interaction. For METH at 0.5 mg/kg, statistical analysis revealed a significant effect of the drug (F 1,16 = 12.41, P < 0.01), of day (F 2,32 = 6.80, P < 0.01) but not effect of environment and no significant interaction between environment and day. For METH at 1.0 mg/kg, statistical analysis revealed a significant effect of the drug (F 1,14 = 41.64, P < 0.0001) but not effect of environment. Only the drug × day interaction reached statistical significance (F 2,28 = 3.34, P = 0.050).

Activating effects of METH in mice reared in standard (SE) or enriched (EE) environments. Locomotor activity in the CPP apparatus after injections of saline and METH during conditioning sessions in saline controls (a), in mice conditioned to METH 0.5 mg/kg (b) and in mice conditioned to METH 1.0 mg/kg (c). Note that for saline control groups (a) locomotor activity in only one compartment is shown. Two-way ANOVA for repeated measures followed by post-hoc Fischer’s PLSD test: * and ** significantly different from saline control level
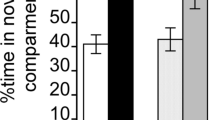
METH induced a dose-dependent CPP that was similar in SE and EE mice (approximately 170 s at METH 0.5 mg/kg and 350 s at METH 1.0 mg/kg) (Fig. 2). Statistical analysis revealed a significant effect of dose (F 2,47 = 7.03, P < 0.01) but not of environment nor a significant dose × environment interaction.

Rewarding effects of METH in mice reared in standard (SE) or enriched (EE) environments. EE and SE mice were tested in the conditioned place preference paradigm (CPP) after conditioning to METH (0.5 and 1 mg/kg i.p.) or saline. EE mice (= open bar) showed similar CPP to METH (1 mg/kg) than SE mice (black bars). n = 8–11 per group. Two-way ANOVA followed by post-hoc Student–Newman–Keuls’s test: ** P < 0.01 different from saline control
EE Exposure has no Impact on METH-Induced Body Temperature Changes
Since METH-induced changes in body temperature may participate in its toxicity (Cadet et al. 2003), we measured core body temperature of mice reared in SE or EE immediately before the first injection (t = 0) and then at time points corresponding to 1 h after each injection (t = 1, 3, 5 and 7 h) of METH (10 mg/kg) or saline (Fig. 3). At the beginning of the experiments, body temperature of EE mice was higher than that of SE mice (37.4 vs. 37.9°C; Student’s t-test DF = 33; T = −2.2; P < 0.05). Measurements in additional groups of mice indicate that this effect was due to the change of location from housing room to experimental room that increased body temperature in EE mice but not SE mice. In fact, body temperature in the housing room was similar in SE and EE mice (approximately 37°C) but increased to about 38°C in EE but not in SE mice (data not shown). In SE and EE mice treated with saline, temperature decreased over time and was similar 3 h after the first injection. In SE mice treated with METH, temperature increased of approximately 1°C reaching its maximum at t = 3 h and decreased to control levels at t = 7 h. In EE mice, temperature did not increase compared to initial levels but stayed elevated, with levels comparable to SE mice treated with METH, until t = 7 h when it decreased to control levels. Statistical analysis revealed a significant effect of treatment (F 1,31 = 6.96, P < 0.05), of time (F 3,93 = 26.01, P < 0.0001), and a significant treatment × time interaction (F 3,93 = 6.6, P < 0.01) but no effect of environment.

Effects of standard (SE) or enriched (EE) environment exposure on METH-induced hyperthermia. Quantitative data represent the changes in temperature in °C after METH (4 × 10 mg/kg) or saline administration to SE or EE mice. Arrows indicate the times of i.p. injections of either saline or METH. METH caused significant increases in body temperature in SE mice and maintained the temperature of EE mice at a higher level than that of the saline-treated mice. n = 8–10 per group. Two-way ANOVA followed by post-hoc Student–Newman–Keuls’s test: * P < 0.05 and ** P < 0.01 different from corresponding time-point in saline controls
EE Exposure does not Prevent METH-Induced Dopamine Neurotoxicity
To investigate the impact of environment on METH-induced decreases in the striatal levels of dopamine and TH, we injected SE or EE mice with four doses of METH 10 mg/kg administered at 2 h intervals (Davidson et al. 2001; Thiriet et al. 2005). Twenty-one days after METH administration, we measured dopamine/DOPAC tissue levels by HPLC and TH levels by western blotting. These measures are often used as two indices of the impact of METH on dopaminergic function (Davidson et al. 2001). We found that METH produced decreases in dopamine and DOPAC striatal concentration (30% approximately) in comparison to saline-treated animals in both SE and EE mice (Fig. 4a). Statistical analysis revealed a significant effect of treatment (F 1,29 = 25.6, P < 0.0001 for dopamine and F 1,29 = 21.9, P < 0.0001 for DOPAC) but no effect of environment and no significant treatment × environment interaction. These results suggest that EE exposure could not counteract the neurotoxic effects of METH.

Dopamine and DOPAC levels in the striatum of mice reared in standard (SE) or enriched (EE) environments and treated with METH. Two dosages of METH were used 10 mg/kg (a) and 5 mg/kg (b). Values represent means ± SEM (nanograms per milligram of tissue weight) of 8–9 mice per group. There were no significant differences in dopamine and DOPAC levels between the saline-injected SE or EE mice. The administration of METH induced a decrease in the dopamine and DOPAC (around 30%) at the two dosages and in both SE and EE mice. Two-way ANOVA followed by post-hoc Fischer’s PLSD test: * P < 0.5 and ** P < 0.01 different from saline controls
To ascertain that the lack of neuroprotection by EE was not due to a ceiling effect of high doses of METH, we then administered a lower METH dosage (4 × 5 mg/kg). Even at this dosage, EE exposure was unable to block METH-induced decreases in dopamine and DOPAC concentrations (Fig. 4b). Statistical analysis revealed a significant effect of treatment (F 1,20 = 22.6, P < 0.0001 for dopamine and F 1,20 = 54.5, P < 0.0001 for DOPAC) but still no effect of environment and no significant treatment × environment interaction.
In line with the lack of effects of EE on dopamine and DOPAC levels, we observed that METH decreased the levels of striatal TH (30–40% approximately) in mice raised either in SE or EE (Fig. 5). Statistical analysis revealed a significant effect of treatment (F 1,31 = 6.96, P < 0.05), of time (F 3,93 = 26.01, P < 0.0001) and a significant treatment × time interaction (F 3,93 = 6.6, P < 0.01) but no effect of environment.

Levels of TH protein in the striatum of mice reared in standard (SE) or enriched (EE) environments and treated with METH. Mice were injected with saline or METH (4 × 10 mg/kg) and were killed 21 days later. SE and EE mice show similar basal levels of TH. Administration of METH induced a decrease of the TH tissue concentration of about 30% in both SE and EE mice. n = 8–9 per group. Two-way ANOVA followed by post-hoc Fischer’s PLSD test: ** P < 0.01 and *** P < 0.001 different from saline controls
METH-Induced Cell Death is not Decreased by Exposure to EE
We then investigated whether EE could have protective effects against the METH-induced apoptosis in the striatum. Because METH acts mainly on DA terminals (Fleckenstein et al. 2000), we focussed our attention on the striatal brain area, which is the main projecting area for DA neurons and where cell death has been previously described (Deng et al. 2001; Thiriet et al. 2005; Zhu et al. 2005). Animals (n = 6–9 per group) were sacrificed 3 days after the injections, a time point where apoptotic processes are at the maximum (Deng et al. 1999). No or very few (<2) TUNEL-positive nuclei were detected in the striatum of saline-treated animals (Fig. 6a, left pictures). After METH injections, positively labelled cells were detected in the striatum of both mice raised in SE or EE indicating that METH induced cell death (Fig. 6a, right pictures). METH induced a significant increase in the number of apoptotic cells; however, no difference was observed between SE and EE mice (Fig. 6b). Statistical analysis revealed a significant effect of treatment (F 1,26 = 25.7, P < 0.0001) but no effect of environment and no significant treatment × environment interaction.

METH-induced striatal cell death in the striatum of mice reared in standard (SE) or enriched (EE) environments. a Representative photomicrographs illustrating the effects of saline or METH (4 × 10 mg/kg) injections on cell death in the striatum of SE and EE mice. Positively labelled-cells undergoing cell death are pointed with arrows (Scale bar = 50 μm). b Quantitative data comparing the apoptotic response of cells to METH or saline in the two groups of mice (SE, left or EE, right). Significantly more TUNEL-positive neurons were observed in mice injected with METH in both SE and EE. n = 6–9 per group. Two-way ANOVA followed by post-hoc Fischer’s PLSD test: ** P < 0.01 and *** P < 0.001 different from saline controls
Discussion
EE has significant neuroprotective effects in a wide range of psychiatric and neurological pathologies (Laviola et al. 2008). Here, we investigated the potential beneficial effects of EE against the rewarding and neurotoxic effects of METH. We found that EE were not able to reduce the ability of METH to induce CPP, decreases of DA and TH striatal levels and to induce striatal cell death. These results suggest that whereas positive life conditions during early stages of life can reduce the risks to develop addiction to cocaine and heroin (Solinas et al. 2008; El Rawas et al. 2009) or provide protection against neurotoxins such as MPTP (Bezard et al. 2003); they are not able to reduce the risks associated with METH use. Therefore, METH appears to be an extremely dangerous substance of abuse and its popularity should raise concerns in our societies.
EE have been previously shown to be able to reduce the activating and rewarding effects of drugs of abuse such as cocaine or heroin in setups similar to those used in this study (Solinas et al. 2008; El Rawas et al. 2009). Therefore, we expected that EE would also reduce the activating and rewarding effects of METH. In contrast, we found that METH induced similar levels of CPP in SE and EE mice. Our CPP results are in agreement with those of Gehrke et al. showing that EE rats and rats reared in social isolation (IE) and pre-treated with saline develop similar preference for the compartment associated to 0.3 and 1 mg/kg METH (Gehrke et al. 2006). They also showed that 1 week after toxic regimen of METH, EE rats show reduced CPP to 0.3 mg/kg of METH but similar CPP to 1 mg/kg of METH. Concerning METH-induced locomotor activity, Gehrke et al. found that the acute effects of low (0.3 mg/kg), but not high (1.0 mg/kg), doses of METH were lower in EE versus IE rats and that, upon the third injection, the effects of high (1.0 mg/kg), but not low (0.3 mg/kg) doses of METH were lower in EE versus IE rats (Gehrke et al. 2006). Our locomotor activity results are only partially in agreement with those results but in total agreement with our CPP results because we found that the effects of METH were always similar between SE and EE mice. The differences between the two studies could be due to species differences or to the fact that Gehrke et al. used social isolation and we used SE as controls. Notwithstanding these minor differences, altogether these results suggest that the vulnerability to develop METH addiction is not reduced by EE. However, it should be considered that the effects of EE on CPP have been shown on some occasions to be at odd with results of intravenous self-administration and in addition, new models of addiction have been developed to better mimic human pathology (Ahmed and Koob 1998; Deroche-Gamonet et al. 2004; Vanderschuren and Everitt 2004). Future studies will be needed to draw more definite conclusions on whether EE influence the vulnerability to METH addiction.
Although METH and cocaine have the same main molecular target, namely the DAT (Fleckenstein et al. 2000), the different results obtained for METH in this study and that of Gehrke (Gehrke et al. 2006) and for cocaine in previous studies (Solinas et al. 2008, 2009) could be explained by the fact that they have different mechanisms of actions within the synaptic cleft. Indeed, cocaine mainly inhibits the reuptake of dopamine by presynaptic neurons, whereas amphetamines, such as METH, additionally promote large release of dopamine by terminals (Fleckenstein et al. 2000). In fact, in the synapse, METH exerts multiple pharmacological effects via two main molecular processes: (1) it stimulates the redistribution of dopamine, serotonin and noradrenaline from storage vesicles to the cytosol; (2) it reverses the transport of these neurotransmitters through plasma membrane transporters (Barr et al. 2006). Consequently, as measured by microdialysis, the increased concentration of extracellular DA is much higher with amphetamines than with cocaine (Di Chiara and Imperato 1988). Because EE mice have lower DAT striatal levels (Bezard et al. 2003), it could have been expected that the effects of METH would be lower in these mice. However, it should be noted that we have already found that EE mice do not differ from SE mice in cocaine-induced elevations of dopamine extracellular levels in the ventral and dorsal striatum (Solinas et al. 2008). Indeed, we proposed that differential reactivity of post-synaptic transduction signal cascade could be a critical mechanism responsible for the protective effects of EE on cocaine-induced CPP and sensitization (Solinas et al. 2008). The present results may indicate that these and/or other mechanisms such as release of glutamate (Kalivas 2007) or norepinephrine (Sofuoglu and Sewell 2009) are overridden by METH.
In our experimental conditions, exposure to EE did not have any beneficial impact against the METH-induced decreases in the levels of dopaminergic markers or cell death at two different toxic regimens of METH. These results are in agreement with those of Gehrke et al. (2006) describing that METH-induced decreases of striatal dopamine levels were not modulated by EE in comparison to rats reared in isolated environments. The lack of beneficial effects of EE is somewhat unexpected because of the previously described neuroprotective impact of EE against several other neurotoxic processes affecting neurons (Laviola et al. 2008). For example, in models of epilepsy or stroke, EE inhibit cell death (van Praag et al. 2000). Transgenic mouse models of Huntington’s neurodegenerative chorea, reared in EE present a slower onset and progression of motor symptoms and cognitive deficits (Laviola et al. 2008). In models of Parkinson’s disease using MPTP (Burns et al. 1985; Jenner and Marsden 1986; Bezard et al. 2003) or 6-hydroxydopamine (6-OHDA) (Anastasia et al. 2009), exposure to EE limited the toxins-induced deleterious effects. Although METH, MPTP and 6-OHDA are all believed to produce neurotoxic effects by stimulating the formation of reactive oxygen species and consequently generating oxidative stress (Smeyne and Jackson-Lewis 2005; Blandini et al. 2008; Krasnova and Cadet 2009), the lack of protection of EE on METH-induced neurodegenerative processes is probably due to specific mechanisms of action of these toxins. A main difference is that METH preferentially acts on neurons terminals whereas MPTP or 6-OHDA damages preferentially cell bodies (Betarbet et al. 2002). Other studies have found that one manipulation can be neuroprotective against one of these toxins but not the others. For example, the activation of adenosine receptors could prevent the METH-induced toxicity but not the one induced by MPTP (Delle Donne and Sonsalla 1994). On the other hand, the deletion of the nociceptin gene was found to reduce the toxicity of MPTP but not that of METH (Brown et al. 2006).
We focussed our analysis of the toxic effects of METH on the striatum the main projection area of dopamine neurons and the area with high levels of DAT, the main molecular target of METH (Jaber et al. 1997; Riddle et al. 2005, 2006). METH produces most of his behavioural, neurochemical and molecular effects by binding to the DAT and releasing massive amounts of dopamine in this region (Fleckenstein et al. 2000). In addition, we have shown that exposure to EE produce plastic changes in this region (Bezard et al. 2003; Thiriet et al. 2008; Solinas et al. 2009) that may be responsible for the reduced reactivity to drug of abuse found in EE mice (El Rawas et al. 2009; Solinas et al. 2009). Therefore, this area appeared the logical starting point for our investigation. On the other hand, METH acts and may produce toxic effects in other brain regions including the prefrontal cortex (Fantegrossi et al. 2008), the olfactory bulb (Deng et al. 2007) and the substantia nigra (Harvey et al. 2009). Whereas future research may be needed to investigate the effects of EE on METH-induced toxicity in these areas, given the lack of difference between EE and SE in the effect of METH on a number of behavioural, neurochemical and molecular measures, we believe that it is unlikely that METH-induced toxicity in these regions would be different between EE and SE.
It is generally considered that the hyperthermia induced by METH participates to the toxicity for monoaminergic systems and striatal neurons (Cadet et al. 2003, for review) because environmental or pharmacological manipulations that protect against METH toxicity have also been shown to prevent or attenuate METH-induced hyperthermia (Ali et al. 1994; Bowyer et al. 1994). However, other reports have indicated that METH-induced hyperthermia and monoaminergic toxicity can be dissociated (Battaglia et al. 2002; Halladay et al. 2003; Thiriet et al. 2005). The latter possibility is supported by studies describing that METH-induced hyperthermia was not affected by genetic manipulations that either exacerbate (Deng et al. 1999) or protect against (Deng et al. 2002) neuronal apoptosis. It has also been suggested that increases in body temperature is the consequence rather than the cause of drugs’ effects in the brain (Brown et al. 2003). In this study, we found that the magnitude of the hyperthermia in comparison to saline-injected animals was similar in EE and SE mice although the initial body temperature of EE mice before any injection was always higher than that of SE mice. We observed that this difference was related to a specific reaction of EE mice to the transfer from the housing room to the experimental room (data not shown). It appears that the higher initial temperature in EE or the higher increase in temperature observed in SE did not influence the toxicity of METH for the dopamine terminals or the apoptosis of striatal cells.
In conclusion, our results indicate that exposure to EE did not provide any protection against the rewarding and neurotoxic effects of METH. Knowing that environmental enrichment provides major beneficial effects against other types of brain injuries, these results indicate that the mechanisms triggered by METH in the brain are highly toxic. Therefore, the increasing popularity of this drug of abuse especially amongst young adults represents a serious public health problem that should be considered with attention.
References
Ahmed SH, Koob GF (1998) Transition from moderate to excessive drug intake: change in hedonic set point. Science 282:298–300
Ali SF, Newport GD, Holson RR, Slikker W Jr, Bowyer JF (1994) Low environmental temperatures or pharmacologic agents that produce hypothermia decrease methamphetamine neurotoxicity in mice. Brain Res 658:33–38
Anastasia A, Torre L, de Erausquin GA, Masco DH (2009) Enriched environment protects the nigrostriatal dopaminergic system and induces astroglial reaction in the 6-OHDA rat model of Parkinson’s disease. J Neurochem 109:755–765
Aron JL, Paulus MP (2007) Location, location: using functional magnetic resonance imaging to pinpoint brain differences relevant to stimulant use. Addiction 102(Suppl 1):33–43
Barr AM, Panenka WJ, MacEwan GW, Thornton AE, Lang DJ, Honer WG, Lecomte T (2006) The need for speed: an update on methamphetamine addiction. J Psychiatry Neurosci 31:301–313
Battaglia G, Fornai F, Busceti CL, Aloisi G, Cerrito F, De Blasi A, Melchiorri D, Nicoletti F (2002) Selective blockade of mGlu5 metabotropic glutamate receptors is protective against methamphetamine neurotoxicity. J Neurosci 22:2135–2141
Betarbet R, Sherer TB, Greenamyre JT (2002) Animal models of Parkinson’s disease. Bioessays 24:308–318
Bezard E, Dovero S, Belin D, Duconger S, Jackson-Lewis V, Przedborski S, Piazza PV, Gross CE, Jaber M (2003) Enriched environment confers resistance to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and cocaine: involvement of dopamine transporter and trophic factors. J Neurosci 23:10999–11007
Blandini F, Armentero MT, Martignoni E (2008) The 6-hydroxydopamine model: news from the past. Parkinsonism Relat Disord 14(Suppl 2):S124–S129
Bowyer JF, Davies DL, Schmued L, Broening HW, Newport GD, Slikker W Jr, Holson RR (1994) Further studies of the role of hyperthermia in methamphetamine neurotoxicity. J Pharmacol Exp Ther 268:1571–1580
Brown PL, Wise RA, Kiyatkin EA (2003) Brain hyperthermia is induced by methamphetamine and exacerbated by social interaction. J Neurosci 23:3924–3929
Brown JM, Gouty S, Iyer V, Rosenberger J, Cox BM (2006) Differential protection against MPTP or methamphetamine toxicity in dopamine neurons by deletion of ppN/OFQ expression. J Neurochem 98:495–505
Burns RS, LeWitt PA, Ebert MH, Pakkenberg H, Kopin IJ (1985) The clinical syndrome of striatal dopamine deficiency. Parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). N Engl J Med 312:1418–1421
Cadet JL, Jayanthi S, Deng (2003) Speed kills: cellular and molecular bases of methamphetamine-induced nerve terminal degeneration and neuronal apoptosis. FASEB J 17:1775–1788
Carroll ME, Anker JJ, Perry JL (2009) Modeling risk factors for nicotine and other drug abuse in the preclinical laboratory. Drug Alcohol Depend 104(Suppl 1):S70–S78
Chang L, Alicata D, Ernst T, Volkow N (2007) Structural and metabolic brain changes in the striatum associated with methamphetamine abuse. Addiction 102(Suppl 1):16–32
Davidson C, Gow AJ, Lee TH, Ellinwood EH (2001) Methamphetamine neurotoxicity: necrotic and apoptotic mechanisms and relevance to human abuse and treatment. Brain Res Brain Res Rev 36:1–22
Delle Donne KT, Sonsalla PK (1994) Protection against methamphetamine-induced neurotoxicity to neostriatal dopaminergic neurons by adenosine receptor activation. J Pharmacol Exp Ther 271:1320–1326
Deng X, Cadet JL (2000) Methamphetamine-induced apoptosis is attenuated in the striata of copper-zinc superoxide dismutase transgenic mice. Brain Res Mol Brain Res 83:121–124
Deng X, Ladenheim B, Tsao LI, Cadet JL (1999) Null mutation of c-fos causes exacerbation of methamphetamine-induced neurotoxicity. J Neurosci 19:10107–10115
Deng X, Wang Y, Chou J, Cadet JL (2001) Methamphetamine causes widespread apoptosis in the mouse brain: evidence from using an improved TUNEL histochemical method. Brain Res Mol Brain Res 93:64–69
Deng X, Jayanthi S, Ladenheim B, Krasnova IN, Cadet JL (2002) Mice with partial deficiency of c-Jun show attenuation of methamphetamine-induced neuronal apoptosis. Mol Pharmacol 62:993–1000
Deng X, Ladenheim B, Jayanthi S, Cadet JL (2007) Methamphetamine administration causes death of dopaminergic neurons in the mouse olfactory bulb. Biol Psychiatry 61:1235–1243
Deroche-Gamonet V, Belin D, Piazza PV (2004) Evidence for addiction-like behavior in the rat. Science 305:1014–1017
Di Chiara G, Imperato A (1988) Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA 85:5274–5278
Diamond MC, Ingham CA, Johnson RE, Bennett EL, Rosenzweig MR (1976) Effects of environment on morphology of rat cerebral cortex and hippocampus. J Neurobiol 7:75–85
El Rawas R, Thiriet N, Lardeux V, Jaber M, Solinas M (2009) Environmental enrichment decreases the rewarding but not the activating effects of heroin. Psychopharmacology (Berl) 203:561–570
Fantegrossi WE, Ciullo JR, Wakabayashi KT, De La Garza R II, Traynor JR, Woods JH (2008) A comparison of the physiological, behavioral, neurochemical and microglial effects of methamphetamine and 3,4-methylenedioxymethamphetamine in the mouse. Neuroscience 151:533–543
Fleckenstein AE, Gibb JW, Hanson GR (2000) Differential effects of stimulants on monoaminergic transporters: pharmacological consequences and implications for neurotoxicity. Eur J Pharmacol 406:1–13
Gehrke BJ, Cass WA, Bardo MT (2006) Monoamine-depleting doses of methamphetamine in enriched and isolated rats: consequences for subsequent methamphetamine-induced hyperactivity and reward. Behav Pharmacol 17:499–508
Halladay AK, Kusnecov A, Michna L, Kita T, Hara C, Wagner GC (2003) Relationship between methamphetamine-induced dopamine release, hyperthermia, self-injurious behaviour and long term dopamine depletion in BALB/c and C57BL/6 mice. Pharmacol Toxicol 93:33–41
Harvey BK, Chou J, Shen H, Hoffer BJ, Wang Y (2009) Diadenosine tetraphosphate reduces toxicity caused by high-dose methamphetamine administration. Neurotoxicology 30:436–444
Jaber M, Jones S, Giros B, Caron MG (1997) The dopamine transporter: a crucial component regulating dopamine transmission. Mov Disord 12:629–633
Jenner P, Marsden CD (1986) The actions of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in animals as a model of Parkinson’s disease. J Neural Transm Suppl 20:11–39
Kalivas PW (2007) Cocaine and amphetamine-like psychostimulants: neurocircuitry and glutamate neuroplasticity. Dialogues Clin Neurosci 9:389–397
Kempermann G, Kuhn HG, Gage FH (1997) Genetic influence on neurogenesis in the dentate gyrus of adult mice. Proc Natl Acad Sci USA 94:10409–10414
Krasnova IN, Cadet JL (2009) Methamphetamine toxicity and messengers of death. Brain Res Rev 60:379–407
Laviola G, Hannan AJ, Macri S, Solinas M, Jaber M (2008) Effects of enriched environment on animal models of neurodegenerative diseases and psychiatric disorders. Neurobiol Dis 31:159–168
Layer RT, Bland LR, Skolnick P (1993) MK-801, but not drugs acting at strychnine-insensitive glycine receptors, attenuate methamphetamine nigrostriatal toxicity. Brain Res 625:38–44
Matuszewich L, Yamamoto BK (2004) Chronic stress augments the long-term and acute effects of methamphetamine. Neuroscience 124:637–646
Nithianantharajah J, Hannan AJ (2006) Enriched environments, experience-dependent plasticity and disorders of the nervous system. Nat Rev Neurosci 7:697–709
Nithianantharajah J, Barkus C, Murphy M, Hannan AJ (2008) Gene-environment interactions modulating cognitive function and molecular correlates of synaptic plasticity in Huntington’s disease transgenic mice. Neurobiol Dis 29:490–504
Nordahl TE, Salo R, Leamon M (2003) Neuropsychological effects of chronic methamphetamine use on neurotransmitters and cognition: a review. J Neuropsychiatry Clin Neurosci 15:317–325
O’Callaghan JP, Miller DB (1994) Neurotoxicity profiles of substituted amphetamines in the C57BL/6J mouse. J Pharmacol Exp Ther 270:741–751
Rampon C, Jiang CH, Dong H, Tang YP, Lockhart DJ, Schultz PG, Tsien JZ, Hu Y (2000) Effects of environmental enrichment on gene expression in the brain. Proc Natl Acad Sci USA 97:12880–12884
Riddle EL, Fleckenstein AE, Hanson GR (2005) Role of monoamine transporters in mediating psychostimulant effects. AAPS J 7:E847–E851
Riddle EL, Fleckenstein AE, Hanson GR (2006) Mechanisms of methamphetamine-induced dopaminergic neurotoxicity. AAPS J 8:E413–E418
Rosenzweig MR, Bennett EL (1996) Psychobiology of plasticity: effects of training and experience on brain and behavior. Behav Brain Res 78:57–65
Scott JC, Woods SP, Matt GE, Meyer RA, Heaton RK, Atkinson JH, Grant I (2007) Neurocognitive effects of methamphetamine: a critical review and meta-analysis. Neuropsychol Rev 17:275–297
Smeyne RJ, Jackson-Lewis V (2005) The MPTP model of Parkinson’s disease. Brain Res Mol Brain Res 134:57–66
Sofuoglu M, Sewell RA (2009) Norepinephrine and stimulant addiction. Addict Biol 14:119–129
Solinas M, Chauvet C, Thiriet N, El Rawas R, Jaber M (2008) Reversal of cocaine addiction by environmental enrichment. Proc Natl Acad Sci USA 105:17145–17150
Solinas M, Thiriet N, El Rawas R, Lardeux V, Jaber M (2009) Environmental enrichment during early stages of life reduces the behavioral, neurochemical, and molecular effects of cocaine. Neuropsychopharmacology 34:1102–1111
Stairs DJ, Bardo MT (2009) Neurobehavioral effects of environmental enrichment and drug abuse vulnerability. Pharmacol Biochem Behav 92:377–382
Tata DA, Raudensky J, Yamamoto BK (2007) Augmentation of methamphetamine-induced toxicity in the rat striatum by unpredictable stress: contribution of enhanced hyperthermia. Eur J Neurosci 26:739–748
Thiriet N, Deng X, Solinas M, Ladenheim B, Curtis W, Goldberg SR, Palmiter RD, Cadet JL (2005) Neuropeptide Y protects against methamphetamine-induced neuronal apoptosis in the mouse striatum. J Neurosci 25:5273–5279
Thiriet N, Amar L, Toussay X, Lardeux V, Ladenheim B, Becker KG, Cadet JL, Solinas M, Jaber M (2008) Environmental enrichment during adolescence regulates gene expression in the striatum of mice. Brain Res 1222:31–41
van Praag H, Kempermann G, Gage FH (2000) Neural consequences of environmental enrichment. Nat Rev Neurosci 1:191–198
Vanderschuren LJ, Everitt BJ (2004) Drug seeking becomes compulsive after prolonged cocaine self-administration. Science 305:1017–1019
Volz TJ, Fleckenstein AE, Hanson GR (2007) Methamphetamine-induced alterations in monoamine transport: implications for neurotoxicity, neuroprotection and treatment. Addiction 102(Suppl 1):44–48
Wilson JM, Kalasinsky KS, Levey AI, Bergeron C, Reiber G, Anthony RM, Schmunk GA, Shannak K, Haycock JW, Kish SJ (1996) Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nat Med 2:699–703
Zhu JP, Xu W, Angulo JA (2005) Disparity in the temporal appearance of methamphetamine-induced apoptosis and depletion of dopamine terminal markers in the striatum of mice. Brain Res 1049:171–181
Acknowledgements
We thank Anne Cantereau for technical assistance with confocal images. We acknowledge the National Institute on Drug Abuse for generous gift of METH. This study was funded by the CNRS, University of Poitiers and the Contrat de Projet Etat Region (CPER) #5.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Thiriet, N., Gennequin, B., Lardeux, V. et al. Environmental Enrichment does not Reduce the Rewarding and Neurotoxic Effects of Methamphetamine. Neurotox Res 19, 172–182 (2011). https://doi.org/10.1007/s12640-010-9158-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-010-9158-2