
Abstract
Purpose
To review and discuss the epidemiology, contributing factors, and approach to clinical management of disorders of sodium and water balance in hospitalized patients.
Source
An electronic search of the MEDLINE, Embase, and Cochrane Central Register of Controlled Trials databases and a search of the bibliographies of all relevant studies and review articles for recent reports on hyponatremia and hypernatremia with a focus on critically ill patients.
Principal findings
Disorders of sodium and water balance are exceedingly common in hospitalized patients, particularly those with critical illness and are often iatrogenic. These disorders are broadly categorized as hypo-osmolar or hyper-osmolar, depending on the balance (i.e., excess or deficit) of total body water relative to total body sodium content and are classically recognized as either hyponatremia or hypernatremia. These disorders may represent a surrogate for increased neurohormonal activation, organ dysfunction, worsening severity of illness, or progression of underlying chronic disease. Hyponatremic disorders may be caused by appropriately elevated (volume depletion) or inappropriately elevated (SIADH) arginine vasopressin levels, appropriately suppressed arginine vasopressin levels (kidney dysfunction), or alterations in plasma osmolality (drugs or body cavity irrigation with hypotonic solutions). Hypernatremia is most commonly due to unreplaced hypotonic water depletion (impaired mental status and/or access to free water), but it may also be caused by transient water shift into cells (from convulsive seizures) and iatrogenic sodium loading (from salt intake or administration of hypertonic solutions).
Conclusion
In hospitalized patients, hyponatremia and hypernatremia are often iatrogenic and may contribute to serious morbidity and increased risk of death. These disorders require timely recognition and can often be reversed with appropriate intervention and treatment of underlying predisposing factors.
Résumé
Objectif
Passer en revue et discuter l’épidémiologie, les facteurs contributifs et l’approche à la prise en charge des troubles de l’équilibre hydrosodé chez les patients hospitalisés.
Source
Nous avons effectué une recherche électronique des bases de données MEDLINE, Embase et de Cochrane Central Register of Controlled Trials et une recherche des bibliographies de toutes les études pertinentes et articles de synthèse pour les comptes-rendus récents traitant de l’hyponatrémie et de l’hypernatrémie, en concentrant notre attention sur les patients en phase critique.
Constatations principales
Les troubles de l’équilibre hydrosodé, excessivement communs chez les patients hospitalisés, et particulièrement ceux en phase critique, sont en général iatrogéniques. Ces troubles sont généralement catégorisés comme hypo-osmolaires ou hyper-osmolaires, selon l’équilibre (c.-à-d. l’excès ou le déficit) d’eau corporelle totale par rapport au contenu sodé corporel total. Les troubles sont traditionnellement reconnus en tant que soit hyponatrémie ou hypernatrémie. Ces troubles pourraient être la manifestation d’une activation neurohormonale accrue, d’un dysfonctionnement organique, d’une évolution défavorable de la maladie ou de la progression d’une maladie chronique sous-jacente. Les troubles hyponatrémiques peuvent être provoqués par des niveaux d’arginine-vasopressine adéquatement élevés (déplétion volumique) ou inadéquatement élevés (syndrome d’antidiurèse inappropriée), des niveaux d’arginine-vasopressine adéquatement supprimés (dysfonctionnement hépatique) ou des altérations de l’osmolarité plasmatique (médicaments ou irrigation des cavités corporelles par des solutions hypotoniques). L’hypernatrémie est la plupart du temps provoquée par une déplétion d’eau hypotonique non remplacée (état mental aggravé et/ou accès libre à de l’eau), mais elle peut également être causée par une translation provisoire de l’eau dans les cellules (à partir de convulsions) et de charge sodée iatrogénique (de l’apport sodique ou par l’administration des solutions hypertoniques).
Conclusion
Chez les patients hospitalisés, l’hyponatrémie et l’hypernatrémie sont souvent iatrogéniques et pourraient contribuer à une morbidité grave et un risque accrue de décès. Ces troubles nécessitent une identification rapide et peuvent souvent être soignés grâce à une intervention adaptée et au traitement des facteurs prédisposants sous-jacents.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Disorders of sodium and water balance are commonly encountered in critically ill patients.1 Critical illness, multi-organ dysfunction, fluid resuscitation, and the numerous additional interventions received routinely by patients admitted to the intensive care unit can interfere with the complex mechanisms that maintain total body sodium and water homeostasis.2
Disorders of sodium and water balance are generally categorized as either hypo-osmolar or hyper-osmolar, depending on the balance (i.e., excess or deficit) of total body water relative to total body sodium content. As sodium is the primary extracellular constituent of serum osmolality, disorders of sodium and water balance can classically be recognized as hyponatremia and hypernatremia. Both of these disorders can contribute to substantial morbidity and mortality, and given their prevalence in critically ill patients, clinicians need to have a solid understanding of their pathophysiology, diagnosis, and management.
Search methodology
In August 2008, we conducted an electronic search of the MEDLINE (inception through August 2008), Embase (inception through August 2008), and Cochrane Central Register of Controlled Trials (inception through August 2008) databases for recent and relevant articles. We also searched the bibliographies of all relevant studies and review articles. Search terms (water balance OR sodium OR hyponatremia OR hypernatremia) were combined with key terms for “outcome” OR “mortality” OR “diagnosis” OR “epidemiology”. The search was limited to studies conducted in humans and reported in English.
Overview of sodium and water homeostasis
Sodium [Na+] is the primary extracellular cation and the most important osmotically active solute in the body. Under normal circumstances, the serum [Na+] is preserved within a fine physiologic range (138–142 mEq l−1) despite large variations in daily sodium and water intake. Sodium metabolism is tightly regulated by the kidney through the interaction of numerous neurohormonal mechanisms, including the renin–angiotensin–aldosterone system, the sympathetic nervous system, and the presence of atrial natriuretic and brain natriuretic peptides. Sodium regulation is closely correlated with the body’s effective circulating volume (ECV), defined as the requisite intravascular volume to provide adequate tissue perfusion. As such, the major determinant of serum [Na+] is in fact the serum water content, and disturbances in sodium balance most often reflect abnormalities in the ECV and serum water content.
Water metabolism, on the other hand, is predominantly regulated by arginine vasopressin (AVP) and is strongly influenced by water intake and output. Arginine vasopressin is produced in the supraoptic and paraventricular hypothalamic nuclei and stored in the posterior pituitary. Arginine vasopressin secretion is tightly regulated by changes in serum osmolality (i.e., as little as 1–2%) detected by osmoreceptors in the anterior hypothalamus and also by changes in mean arterial pressure and/or blood volume detected by baroreceptors in the aortic arch and carotid bodies. Arginine vasopressin controls the water permeability of the kidney by directing the insertion of aquaporin-2 (AQP-2) channels on the luminal surface of the distal tubules and collecting duct. Arginine vasopressin induces an increase in AQP-2 channels and acts to stimulate free water reabsorption and anti-diuresis.
Hyponatremia
Hyponatremia is commonly defined as a serum [Na+] < 135 mmol l−1; however, this definition may vary across different institutional laboratories. The presence of hyponatremia most commonly indicates an underlying disorder of an excess in body water relative to body sodium content. Less commonly, it may result from a depletion of body sodium content in excess of concurrent body water losses.
Epidemiology
Hyponatremia is recognized as the most common electrolyte abnormality encountered in clinical medicine. Its prevalence in the United States is estimated to be between 3.2 and 6.1 million patients/year.3 Approximately 1% of these cases are classified as acute and symptomatic, 4% as acute and asymptomatic, 15–20% as chronic and symptomatic, and 75–80% as chronic and asymptomatic. This prevalence is associated with a considerable burden on health resources, as an estimated 75% of these patients require treatment in hospital.3
Epidemiologic studies have found that hyponatremia occurs in approximately 1–2% of hospitalized patients.4 However, the incidence varies depending on the threshold for diagnosis, and the population assessed. For example, hyponatremia (serum [Na+] ≤ 130 mmol l−1) has been described in 4.4% of patients after surgery5 and in nearly 30% of patients admitted to intensive care (serum [Na+] ≤ 134 mmol l−1).6 A variety of risk factors have been reported for hospital-acquired hyponatremia, including older age, diabetes mellitus, chronic kidney disease, surgery, pulmonary infection, diuretic therapy, administration of antibiotics, opioid analgesia, and the use of hypotonic intravenous fluids.7–9
It is important to recognize that hyponatremia, while frequent, is not a trivial diagnosis. It is associated with serious complications that have been linked to increased morbidity and mortality.1,8,10–13 The presence of hyponatremia after an acute ST-elevation myocardial infarction in congestive heart failure and in patients with cirrhosis has been found to predict mortality.10–13 Similarly, in critically ill patients, severe hyponatremia (serum [Na+] < 125 mmol l−1) has been shown to be an independent predictor of hospital mortality, with an estimated risk for death approaching 40%.1 Gill et al.8 recently found that severe hyponatremia (serum [Na+] < 125 mmol l−1) was associated with significantly higher mortality (27% vs. 9%, P = 0.009) and a longer duration of hospitalization (16 days vs. 13 days, P < 0.005). Moreover, mortality was reported higher for those patients whose hyponatremia initially worsened after hospital admission.8
Clinical presentation of hyponatremia
Symptoms attributable to hyponatremia correlate both with the severity and the rate of decline in serum [Na+] and generally reflect neurologic dysfunction induced by cerebral edema. A reduction in serum [Na+] creates an osmotic gradient that favours water movement into the brain. This increase in brain intracellular volume contributes to cerebral edema and raised intracranial pressure and leads to the appearance of neurologic manifestations.
Mild hyponatremia (serum [Na+] 130–135 mmol l−1) can often be asymptomatic, but with further acute declines in serum [Na+], overt symptoms become more apparent. Non-specific symptoms occur with a serum [Na+] in the range of 120–130 mmol l−1, such as fatigue, malaise, nausea, and unsteadiness. Rapid declines to serum [Na+] < 115–120 can provoke headache, restlessness, lethargy, and obtundity that may progress to seizures, coma, brainstem herniation, respiratory arrest, and death.14
Alternatively, hyponatremia that evolves more gradually (i.e., over days or weeks) may present with a much lower serum [Na+] prior to the development of symptoms. This occurs as a result of the brain undergoing a process of intracellular adaptation to preserve osmotic balance and prevent edema. Throughout hours and days, the brain transports osmoles (i.e., sodium, potassium, chloride) from the intracellular to the extracellular space, followed later by active transport of several organic solutes (i.e., osmolytes), such as glutamine, glutamate, taurine, and myo-inositol. This process aids in maintaining osmotic balance by contributing to early water loss from the brain, which attenuates subsequent hyponatremia-induced brain edema, and hence, leads to a greater threshold decline in serum [Na+] prior to symptoms.
Diagnostic approach
Hyponatremia can be broadly classified, based on serum osmolality, into the categories of hypo-osmolar, iso-osmolar, or hyper-osmolar disorders. The underlying cause of hyponatremia is usually evident after a thorough medical history, a physical examination, and selected serum and urinary tests, particularly, serum osmolality, urine osmolality, and urine [Na+]. The medical history should focus on presence of co-morbid illnesses, acute illnesses, medications, and other therapies or interventions that may predispose to the development of hyponatremia (Table 1). A focused physical examination should provide an estimate of volume status; a reduction in ECV may be suggested by orthostatic changes in heart rate and blood pressure, large variations in pulse pressure, low jugular or central venous pressure, and other surrogates, such as reduced skin turgor, furrowed tongue, and dry mucus membranes, or axillae. On the other hand, an expansion in ECV would be suggested by increased jugular or central venous pressure, pleural effusions, ascites, and peripheral edema.
Hypo-osmolar hyponatremia
Hypo-osmolar hyponatremia is most commonly encountered in critically ill patients and, according to an assessment of the ECV, can generally be classified into hypovolemic, isovolemic, or hypervolemic hyponatremia.
Hypovolemic hypo-osmolar hyponatremia
The simultaneous loss of solute and water from the extracellular space results in a reduced ECV and, in an attempt to restore vascular volume and attenuate free water loss, triggers the non-osmotic release of AVP. Subsequent intake of hypotonic fluids or free water by ingestion or infusion leads to hyponatremia. There are numerous conditions that contribute to true volume depletion, such as insensible fluid loss, gastrointestinal losses, hemorrhage, and renal fluid and solute losses (i.e., diuretics, mineralocorticoid deficiency, chronic nephropathies) (Table 1). This form of hyponatremia has emerged as an important cause of morbidity in endurance athletes. An estimated 13% of runners in the 2002 Boston marathon had serum [Na+] < 135 mmol l−1, and approximately 1% had critical values <120 mmol l−1.15 Moreover, the frequent use of non-steroidal anti-inflammatory drugs may further compound hyponatremia in these athletes. In the absence of exposure to diuretics, true hypovolemia in these patients may be corroborated by demonstration of a urine [Na+] < 10–20 mmol l−1.
Cerebral salt wasting syndrome (CSWS) is a unique disorder of the hypothalamic-renal axis characterized by natriuresis and volume depletion, followed by AVP-induced water retention. This leads to hyponatremia typified by an inappropriately high urine osmolality, a high urine [Na+] generally >40 mEq l−1, and, if measured, an increased serum [AVP]. While the pathogenesis is not completely understood, increased sympathetic nervous system outflow, along with raised levels of atrial and brain natriuretic peptides, may, in part, mediate the inciting natriuresis and volume depletion. Cerebral salt wasting syndrome typically occurs in critically ill patients with intracranial injury, which is often associated with subarachnoid hemorrhage or traumatic brain injury, and is less commonly described after neurosurgical procedures with glioma, tuberculous, or carcinomatous meningitis.16 Cerebral salt wasting syndrome is often difficult to differentiate from the syndrome of inappropriate antidiuretic hormone (SIADH) secretion, which is also common after neurologic injury. The key difference for CSWS is clear evidence of volume depletion and increased urine sodium excretion prior to development of hyponatremia; whereas in SIADH, patients are typically euvolemic or mildly volume expanded.17
Isovolemic hypo-osmolar hyponatremia
There are several important causes of isovolemic hypo-osmolar hyponatremia, including SIADH, endocrinopathies, such as adrenal insufficiency, or hypothyroidism and pregnancy.
The SIADH is characterized by an inappropriate or persistent release of AVP that results in a decreased capacity for free water excretion. This syndrome is the most common cause of acquired hyponatremia in hospitalized patients.4 The diagnostic criteria for SIADH are shown in Table 2. The major criteria for the diagnosis of SIADH are evidence of serum hypo-osmolality (<275 mOsm kg−1) and a less than maximally dilute urine osmolality >100 mOsm kg−1. In addition, patients are euvolemic, have normal acid-base and potassium balance, and a urine [Na+] that is typically >40 mmol l−1. In general, SIADH is a diagnosis of exclusion and can only be confirmed in the context of normal kidney, thyroid, and adrenal function. There are several conditions encountered in critically ill patients that can lead to SIADH. The SIADH can be broadly categorized into disorders of the central nervous system, pulmonary disorders, disorders associated with medications or tumours, and a variety of miscellaneous causes (Table 3). Interestingly, an estimated one-third of patients with SIADH counter the inappropriately elevated AVP by resetting the osmostat downwards to a serum [Na+] typically in the range 125–130 mmol l−1. These patients are often asymptomatic and achieve relative stability in serum [Na+]. As such, confirming the diagnosis is important and can have significant implications for subsequent therapy.
Adrenal insufficiency generally leads to hyponatremia, due to an increased release of AVP and subsequent diminished water excretion. Cortisol deficiency may contribute to reductions in cardiac output and blood pressure, thus stimulating a non-osmotic release of AVP. In addition, AVP is an adrenocorticotropic hormone (ACTH) secretagogue, thus AVP release may be stimulated secondary to increased release of ACTH, due to the lack of negative feedback from absent serum cortisol.18 Similarly, aldosterone deficiency leads to sodium wasting and reductions in ECV stimulating AVP release.
The pathophysiology of hyponatremia in hypothyroidism remains incompletely understood. Studies have suggested these patients have impaired free water excretion due to inability to maximally suppress AVP secretion; however, this may be aggravated by declines in cardiac output that stimulate the non-osmotic release of AVP and by reductions in glomerular filtration that further impair free water clearance.19
During pregnancy, increased serum levels of human chorionic gonadotropin released from the placenta is believed to be associated with a downward reset osmostat (≤5 mmol l−1) leading to mild asymptomatic hyponatremia.20
Hypervolemic hypo-osmolar hyponatremia
There are several conditions that can predispose to hypo-osmolar hyponatremia in the context of an excess of total body water and sodium. Congestive heart failure, cirrhosis, and chronic kidney disease (i.e., nephrotic syndrome) all share a similar pathophysiology for the development of hyponatremia in edematous states.
Congestive heart failure is classically associated with extracellular fluid overload. However, the reductions in cardiac output (and blood pressure) cause a relative reduction in ECV. These hemodynamic changes activate carotid baroreceptors that stimulate the non-osmotic release of AVP. Additionally, the impaired cardiac output contributes to reduced renal perfusion, which, in turn, activates the renin-angiotensin-aldosterone system and sympathetic nervous system, thereby amplifying renal sodium retention and secondarily decreasing free water excretion. These alterations are further exacerbated by concomitant diuretic therapy. However, this maladaptive positive feedback leads to dilutional hyponatremia associated with progressive hypervolemia.21
Advanced cirrhosis is typically characterized by significant splanchnic and systemic vasodilatation. This leads to relative reductions in ECV, non-osmotic release of AVP, and diminished capacity for free water excretion, leading to serum [Na+] of <125–130 mmol l−1 in up to 50% of patients. The increase in AVP secretion, and thus level of hyponatremia, is often proportional to the underlying progression and severity of cirrhosis.22 Diuretic therapy, often used to treat ascites, can worsen hyponatremia in cirrhotic patients by reducing ECV, stimulating compensatory increases in AVP release, and further impairing free water excretion.
In advanced chronic kidney disease (≥stage IV), the reduction in nephron mass and glomerular filtration are associated with progressive impairments in capacity for maximal urine dilution and free water excretion, such that water retention commonly predisposes to hyponatremia.
Primary polydipsia is characterized by an abnormal thirst stimulus leading to an increased and/or excess of free water intake. It is often found in psychiatric illness or with prescription of anti-psychotic medications that cause a dry mouth.23 Infrequently, primary polydipsia can occur from infiltrative diseases of the hypothalamus (i.e., sarcoidosis) that disrupt the normal sensation of thirst.24 Severe hyponatremia has also been described after acute water intoxication in workers undergoing urine drug testing.25 In these circumstances, water intake exceeds the renal capacity for excretion, despite a maximally dilute urine (i.e., osmolality < 100 mOsm kg−1). Ingestion of ecstasy (3,4 methyldioxymethamphetamine or MDMA) has also been associated with severe acute hyponatremia.26 The underlying pathophysiology is thought to result from a large intake of free water coupled with SIADH.
Poor dietary intake of solute can directly impair capacity for free water excretion and lead to dilutional hyponatremia. This may be encountered in chronic alcoholics (i.e., beer potomania) and malnourished patients.27,28 These patients have appropriately dilute urine; however, due to a low intake of solute (i.e., sodium and potassium), the daily solute excretion will decrease to <200–250 mOsm kg−1 (normal 600–900 mOsm kg−1) and lead to a reduction in the maximal achievable urine output.
Hyperosmolar hyponatremia
The accumulation of osmotically active particles in the plasma induces an osmotic efflux of water from the intracellular space to the extracellular space, resulting in both hyponatremia and hyperosmolality. This is often encountered with marked hyperglycemia (i.e., diabetic ketoacidosis, hyperosmolar non-ketotic hyperglycemia) and less commonly with use of mannitol, glycerol, or sorbitol, and the administration of radiocontrast media. Similarly, hyperosmolar hyponatremia has also been described with the use of IVIg suspended in 10% maltose solution.29
The calculation to correct the serum [Na+] for hyperglycemia was shown on average as a decrease of 2.4 mmol l−1 in serum [Na+] per 5.6 mmol l−1 increase in serum [glucose].30 However, this relationship was non-linear and may vary, indicating that this calculation is at best an estimate. Other variables are not factored in, such as ongoing water loss from osmotic diuresis and the influence of insulin administration, both of which will contribute to raising the serum [Na+].
Iso-osmolar hyponatremia
Iso-osmolar hyponatremia can occur with the accumulation of isotonic non-sodium containing fluid to the extracellular space or by marked elevations in serum compounds such as proteins and lipids (i.e., pseudohyponatremia). This has been reported during selected surgical procedures (i.e., transurethral resections of the prostate, bladder tumour resection, and hysteroscopy) that involve the large volume irrigation of closed body spaces with hypotonic glycine or sorbitol-containing solutions (see below).
The normal content of serum is approximately 93% water and 7% non-aqueous substances that principally include proteins and lipids. In general, the non-aqueous proportion of serum does not influence osmolality. However, in disorders causing marked elevations in serum proteins (i.e., multiple myeloma, hypergammaglobulinemia) or lipid content (i.e., hypertriglyceridemia, elevated chylomicrons), the non-aqueous proportion of serum is increased relative to the aqueous portion, leading to an artifactual decrease in serum [Na+] (i.e., pseudohyponatremia) despite no actual change to serum [Na+] or serum osmolality. This problem has largely been overcome by the use of ion-selective electrodes that directly measure serum [Na+].
Hyponatremia in the perioperative period
Hyponatremia after surgery is common; it can often go unrecognized due to clinical overlap with the sequale of postanesthesia, and may contribute to iatrogenic morbidity and mortality.31–33 Observational studies report variable occurrences of early postoperative hyponatremia, largely due to different surgical populations and serum [Na+] thresholds for defining hyponatremia. In general, orthopedic, and gynecologic surgery, the incidence of postoperative serum [Na+] < 135 mmol l−1 has been reported to occur in 2–10% of patients; however, the risk may be modified by illness severity (i.e., acute physiologic stress), and rates of up to 31% have been reported.34–37 More significant hyponatremia (serum [Na+] < 120–130 mmol l−1) is less common and has been reported in the range of 1–5% of patients.35–38
The pathophysiology of postoperative hyponatremia is complex and often multifactorial.39 In general, impaired free water excretion (i.e., reduced renal function, reduced renal tubular dilution capacity, non-osmotic release of AVP), together with continued hypotonic fluid administration or water intake in the perioperative period, contribute to reductions in serum [Na+]. The constellation of clinical contributors may include non-osmotic stimuli for the release of AVP, such as surgical pain, nausea, anxiety, stress, inflammation, and various medications, along with subclinical volume depletion from a prolonged preoperative fast. Although relatively uncommon, a true excess in total body free water, characterized by a positive fluid balance in the postoperative period, coupled with a relatively preserved total body sodium content (i.e., acute water intoxication) is a potentially devastating iatrogenic complication.31,40
Data have accumulated to indicate that women may be at a higher risk than men for acute reductions in serum [Na+] in the immediate postoperative period. In a small observational study, Amede et al.41 found that women undergoing pelvic surgery had acute declines in estrogen, progesterone, and aldosterone, postoperatively. These changes correlated with acute reductions in serum [Na+] and elevated serum AVP levels, despite fluid therapy with only normal saline/Ringer’s lactate and a positive net sodium and fluid balance in the 24 h after surgery. The implications are that women may have a greater tendency to retain free water in response to surgical stress. In a case-control study, Ayus et al.33 found that women and men are equally likely to develop hyponatremia and hyponatremic encephalopathy after surgery; however, menstruant women were 28 times more likely than men or postmenopausal women to die or to suffer permanent neurologic injury. There is speculation that estrogen and/or progesterone may facilitate brain cell adaptation to plasma hypotonicity and that decreased levels may interfere with the normal compensatory decreases in brain cell osmolality that occur in response to changes in extracellular tonicity.33,41
The concept of “sick cell syndrome” may be an under-recognized mechanism of hyponatremia in critically ill and postoperative patients.42–45 Sick cell syndrome is believed to be caused by dysfunctional cell membrane integrity and increased permeability associated with cellular Na+/K+ ATPase pump dysfunction. Loss of membrane integrity may lead to leakage of intracellular solutes that induce an acute increase in extracellular osmolality and predispose to water translocation and redistributive (rather than dilutional) hyponatremia. The redistributive hyponatremia is unrelated to total body free water or sodium balance and may be characterized by an increase in both serum and urine osmolar gap.
Large volume irrigation of closed body spaces with hypotonic solutions can lead to significant perioperative fluid and electrolyte shifts and is a well recognized iatrogenic cause of hyponatremia in patients undergoing transurethral prostatectomy or hysteroscopy. These procedures use large volumes of glycine or sorbitol-containing intra-cavitary irrigating solutions. As a consequence, variable amounts of this fluid can be absorbed, either directly through large prostatic veins or indirectly via leaked fluid in the retroperitoneal space, and can lead to a dilutional reduction in serum [Na+].46 Postoperative decreases in serum [Na+] to <100–110 mmol l−1 have been described and can be associated with serious neurologic sequelae and death.46 Similar problems have been described with use of large volume glycine irrigation during hysteroscopy.47 This diagnosis is supported by the finding of a large serum osmolal gap ≥30–40 mOsm kg−1, whereas a normal serum osmolal gap is ≤5–10 mOsm kg−1. The osmolar gap is determined by the laboratory measure minus the calculated serum osmolality and can be estimated by the equation:
Recently, several series and small randomized trials have shown that large volume saline irrigation with bipolar transurethral resection is equally efficacious and results in a reduced risk of postoperative dysnatremia compared with glycine-based conventional monopolar transurethral resection.48–52
In general, many patients undergoing surgery should be considered at risk for the development of postoperative hyponatremia. Any neurologic symptoms during the perioperative period should raise suspicion for hyponatremia as a contributor and should prompt urgent evaluation of serum [Na+]. Judicious attention to perioperative water intake, fluid therapy, and balance, along with other contributors to hyponatremia can help to avoid the potential consequences of this preventable and often iatrogenic complication.
Principles of clinical management
There are a few essential questions that should be asked in the approach to the clinical management of the patient with hyponatremia:
-
1.
What is the underlying diagnosis of hyponatremia, and, if known, is there an etiology-specific treatment?
-
2.
What rate of serum [Na+] correction is considered safe, given the clinical context?
-
3.
What is the risk of central pontine myelinolysis (osmotic demyelination)?
-
4.
What is the optimal method for raising the serum [Na+]?
-
5.
What is the management approach when the serum [Na+] has been corrected too rapidly?
Underlying diagnosis and etiology-specific treatment
The initial step in managing the patient with hyponatremia is confirming the diagnosis and instituting and/or discontinuing cause-specific predisposing factors. For example, this may include correction of ECV depletion with isotonic saline, discontinuation of diuretic therapy or other medications that may contribute to SIADH; initiation of corticosteroid or thyroid hormone replacement, if deficient; and restriction of free water intake in SIADH or primary polydipsia.
Rate of correction of serum [Na+] and central pontine myelinolysis
The rate of correction of serum [Na+] depends on the clinical presentation and the presence of symptoms.
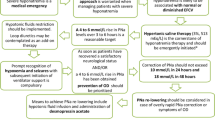
In general, patients presenting with severe symptomatic hyponatremia (i.e., altered mental status, seizures, coma) require urgent therapy to prevent serious neurologic injury (i.e., brain herniation) or death. These patients present with acute severe falls in serum [Na+] (i.e., postoperative, associated with ecstasy, exercise-induced), acute or chronic declines in serum [Na+], where brain adaptation to hyponatremia has already occurred; or they have low tolerance to changes in brain water content (i.e., pre-existing intra-cranial pathology). In these circumstances, where patients manifest symptoms of cerebral edema, a rapid initial correction at a rate of 1.5–2.0 mmol l−1 per hour for 3–4 h is necessary. These patients often receive hypertonic saline to achieve rapid initial rises in serum [Na+]. While those patients with acute declines in serum [Na+] developing in <24 h often tolerate more rapid correction, determination of the onset and duration of hyponatremia prior to presentation is often not possible. Therefore, the rate of correction should ideally be limited to ≤10 mmol l−1 and ≤18 mmol l−1 over the first 24 and 48 h, respectively.53 This course of action requires frequent monitoring of serum [Na+] in response to therapy and continual assessment for over-correction.
In patients with severe hyponatremia (serum [Na+] < 120 mmol l−1) associated with milder symptoms (i.e., nausea, fatigue, lethargy, confusion, unsteadiness), active intervention is warranted; however, there is less urgency than for patients with clinical evidence of cerebral edema. In these symptomatic patients, serum [Na+] can be increased at a rate of 1.0 mmol l−1 per hour for 3–4 h; however, the same true rate of correction over the first 24 and 48 h should be observed.53
For asymptomatic patients, serum [Na+] must be corrected slowly, as cerebral adaptation has occurred at a recommended rate <10–12 mmol l−1 and <18 mmol l−1 per 24 and 48 h, respectively.53
After correction of hyponatremia, the development of central pontine myelinolysis appears most commonly associated with the underlying chronicity of hyponatremia (i.e., allowance for cerebral adaptation) and the rate of rise of serum [Na+] within the first 48 h.54 Sterns et al.54 found no neurologic complications for patients with serum [Na+] corrected at a rate <12 mmol l−1 per 24 h and <18 mmol l−1 per 48 h, or for patients with an initial serum [Na+] < 120 mmol l−1 corrected at a rate ≤0.55 mmol l−1 per hour. Risks for osmotic demyelination may also include hypokalemia, liver disease, poor nutritional state, or burn injury.55 Unfortunately, central pontine myelinolysis has no effective therapy. Also, it generally has a poor prognosis, often characterized by permanent or only partially recovered neurologic deficits. Prevention is paramount.
Methods to raise the serum [N+]
The methods primarily used to raise serum [Na+] are broadly divided into the following categories: fluid restriction, sodium administration, or use of selective vasopressin receptor antagonists.56 The specific therapy prescribed will vary depending on the underlying etiology and its severity.
Fluid restriction with the goal of achieving a negative fluid balance is suitable therapy for primary polydipsia and, in clinical conditions, is associated with either normovolemia (i.e., SIADH) or volume overload (i.e., cirrhosis, congestive heart failure, renal failure).
Administering sodium, most often as saline solution, is appropriate therapy for the hyponatremia associated with volume depletion. A crude estimate of the degree to which 1000 ml of saline solution will raise the serum [Na+] can be calculated by the following formula56:
TBW refers to total body water. Normal total body water for men and women is approximately 0.6 and 0.5 times the lean body weight. However, there are several inherent limitations to this formula. This calculation does not account for ongoing sodium excretion or shifts in total body water and may be inappropriate for use in patients with SIADH due to preserved renal sodium handling.
In SIADH, therapy should initially be directed at reversing the underlying precipitant and fluid restriction. If fluid therapy is needed to rapidly correct the serum [Na+] in SIADH, the total effective osmolality of the fluid administered must exceed the urine osmolality, otherwise the serum [Na+] may further decline. In these circumstances, 0.9% normal saline (0.9% NS) is often ineffective, and a more hypertonic saline solution is needed.
Selective vasopressin receptor antagonists (i.e., tolvaptan, conivaptan) act to inhibit AVP in the renal tubule, to induce an electrolyte-free water diuresis, and to raise serum [Na+].53 For the most part, these drugs have been used for hyponatremia associated with normovolemic or edematous states (i.e., SIADH, heart failure).57–62 While preliminary data are promising that these agents may be alternatives to fluid restriction or saline administration, clinical experience is limited, particularly to support their use in managing acute severe hyponatremia. Vasopressin receptor antagonists are approved by the Food and Drug Administration for use in the United States; however, they are not yet approved for use in Canada.
Management of overly rapid serum [Na+] correction
Excessively rapid correction of hyponatremia can predispose to the development of cerebral edema, central pontine myelinolysis, coma, and death. By identifying patients considered at-risk, attending appropriately to clinical management, and frequent monitoring, these disabling, largely iatrogenic complications are preventable.
There are selected patient cohorts and/or therapies where there may be an increased risk for overly rapid correction. Potentially, physicians should anticipate these clinical contexts where rapid correction may occur. The administration of hypertonic saline to patients with severe symptomatic hyponatremia can contribute to rapid elevations in serum [Na+]. Similarly, rapid correction can occur in hypovolemic patients with hypo-osmolar hyponatremia, where ECV has been restored with 0.9% NS. Rapid correction occurs in response to both administration of 0.9% NS (hypertonic relative to serum) and subsequent AVP suppression after correction of ECV deficit, leading to diuresis of excess free water. Hyponatremia in adrenal insufficiency is associated with elevated AVP secretion that is suppressed after corticosteroid replacement and may result in rapid rises in serum [Na+]. In end-stage kidney disease, rapid correction of hyponatremia can occur during a session of dialysis using a high dialysate [Na+]. In these patients, customization of dialysate solution is needed to ensure a more gradual correction of serum [Na+]. Primary water intoxication (primary polydipsia) is associated with suppressed AVP secretion, and the excess of free water can be rapidly excreted after appropriate therapy with fluid restriction. While these patients have been shown to tolerate rapid rises in serum [Na+] without developing neurologic complications, these findings have not been universal.54,63 Several risk factors have been identified for patients developing neurologic sequelae with overly rapid correction, including hypokalemia, malnutrition, pre-existing alcoholism, and burns.55,63 It has been suggested that an even slower rate of correction of hyponatremia in these patients is desirable.63
Hypernatremia
Hypernatremia is typically defined as a serum [Na+] > 145 mmol l−1; however, this definition may again vary across different laboratories. The presence of hypernatremia, by and large, indicates a relative deficit of body water in relation to body sodium content. Less commonly, it can also be induced by administering an excess of sodium load relative to water.
Epidemiology
Hypernatremia (serum [Na+] > 148 mmol l−1) has been reported to occur in an estimated 1% of hospitalized elderly patients;64,65 yet, this incidence will fluctuate depending on the threshold serum [Na+] and the population being evaluated. Hypernatremia (serum [Na+] ≥ 150 mmol l−1) has been reported in approximately 9% of critically ill patients at the time of admission and has increased by an additional 5.7% of patients during their course in the intensive care unit.66
Several factors have been associated with an increased risk of hypernatremia, including older age, prior brain injury, diabetes mellitus, surgery, diuretic therapy, and altered mental status.64–66 Most hypernatremia is hospital-acquired iatrogenic and can be attributed to inadequate or inappropriate prescription of fluid therapy to patients with identifiable water losses, impaired thirst, or reduced access to free water.65
Similar to hyponatremia, a diagnosis of hypernatremia has been associated with an increased risk for hospital death.64,65,67–69 However, it may be difficult to separate the contribution to mortality of hypernatremia alone from that of the underlying disease process and overall illness severity. Nonetheless, mortality has been shown higher for critically ill patients with hypernatremia (serum [Na+] ≥ 150 mmol l−1). Snyder et al. found hospital mortality for elderly patients with hypernatremia seven-fold higher than for age-matched hospitalized patients.64 In addition, Polderman et al.66 showed that the risk of hospital death was higher for patients with hospital-acquired hypernatremia than for patients with hypernatremia present at the time of admission to the intensive care unit (32% vs. 20%, respectively, P < 0.001). Recently, Hoorn et al.68 showed hypernatremia (serum [Na+] ≥ 150 mmol l−1) was independently associated with higher mortality (48% vs. 10%, P < 0.001). The prognosis for patients presenting with more extreme hypernatremia (serum [Na+] > 180–200 mmol l−1) is poor,70 yet survival has been described,71–73 particularly in children.74 These patients, however, may have residual and/or permanent neurologic disability.75
Clinical presentation of hypernatremia
The clinical manifestations of hypernatremia are principally neurologic and correlate with both the severity and the rapidity of the onset of change in serum [Na+]. The increase in serum [Na+] leads to water movement from the brain intracellular space to the extracellular compartment. Large shifts in brain water content can decrease brain volume and predispose to vascular damage and intracerebral and/or subarachnoid hemorrhage and can potentially lead to irreversible neurologic injury. While typically described as a late complication after rapid correction of hyponatremia, osmotic demyelination has infrequently been described with acute severe hypernatremia.76
Initial symptoms may be subtle and non-specific, including anorexia, restlessness, irritability, lethargy, muscle weakness, and nausea. These can progress to more serious manifestations, such as hyper-reflexia, seizures, and coma. Severe symptoms are generally seen after acute and large elevations to serum [Na+] > 158–160 mmol l−1.77
Elevations in serum [Na+] typically generate an intense sensation of thirst that acts to protect against the neurologic injury associated with severe hypernatremia. This normal physiologic response, however, may be impaired in patients with an altered mental status or with hypothalamic lesions attenuating their sense of thirst (i.e., hypodipsia-adipsia). Older age is also associated with a diminished osmotic stimulation for thirst that may further predispose to reduced capability to replace water loss.78,79
Diagnostic approach
Hypernatremia can be broadly categorized according to the etiologic factors involved, including free water depletion that is unreplaced (i.e., reduced ECV or dehydration), hypodipsia, and an excess intake of sodium or hypertonic solution (i.e., expanded ECV) (Table 4).
The cause of hypernatremia is typically evident from routine history and physical examination; however, additional diagnostic tests of the AVP-renal axis may be needed to establish the diagnosis. In general, an increase in serum [Na+] > 145 mmol l−1 or serum osmolality > 295 mOsm kg−1 H2O represents a potent stimulus for sufficient AVP secretion to cause maximal concentration of urine to >700–800 mOsm kg−1 H2O. Further exogenous AVP administration would likely result in no further increase in urine osmolality. Hypernatremia, in the context of maximally concentrated urine, generally suggests insufficient water intake, hypotonic insensible or gastrointestinal losses, or sodium overload. The measurement of urinary [Na+] may further aid in discriminating a reduced ECV from sodium overload. In conditions of reduced ECV, the urinary [Na+] is typically <25 mEq l−1; whereas, in circumstances of sodium overload, urinary [Na+] is often >100 mEq l−1.
Alternatively, a serum [Na+] > 145 mmol l−1 or serum osmolality > 295 mOsm kg−1 H2O associated with a urine osmolality < 700–800 mOsm kg−1 H2O suggests a defect in the capability for urinary concentration. More specifically, if the urine osmolality is less than the serum osmolality, then a diagnosis of either central (AVP deficiency) or nephrogenic (AVP insensitivity) diabetes insipidus (DI) is confirmed. These can be distinguished by administering exogenous AVP (i.e., dDAVP 10 μg intranasal or vasopressin 5 μg subcutaneous). In central DI, urine osmolality will increase by ≥50%, whereas no significant change will occur in nephrogenic DI.
Insufficient water intake
Hypernatremia, due to inadequate water intake, is usually a consequence of insufficient access to free water, an impaired or altered sensation of thirst, or neurologic injury with alterations to mental status. Inadequate access to free water, particularly in hospitalized patients, is probably more common than appreciated. For instance, in one observational study, 86% of hospitalized patients (mostly elderly) found with hypernatremia had evidence of inadequate access to water.65 In addition, there are likely age-related declines in thirst (i.e., hypodipsia-adipsia). For example, in response to a 24-h water deprivation, despite increases in serum osmolality, serum [Na+], and vasopressin levels, elderly men experienced reduced thirst and water intake when compared with younger adults.78,79
True deficits in thirst and osmoregulation are more likely to occur in patients with acquired hypothalamic structural lesions from conditions such as traumatic brain injury, tumours, granulomatous infiltration (i.e., sarcoid), and vascular disease.80
Conditions that cause an alteration in a patient’s mental status (i.e., delirium) or that bring about a significant neurologic injury (i.e., stroke) would certainly aggravate age-related declines in thirst and hypodipsia from hypothalamic lesions.
Diabetes insipidus
In general, a urine osmolality < 800 mOsm kg−1 H2O, in the setting of an elevated serum osmolality (>295 mOsm kg−1 H2O) or hypernatremia (serum [Na+] > 145 mmol l−1), is indicative of a renal concentrating defect. In the absence of another etiology to account for the high urine osmolality such as osmotic diuresis, this generally reflects the presence of DI.
Central DI refers to polyuria and a urinary concentrating defect as a consequence of a deficiency of AVP secretion from the hypothalamic-pituitary axis. True central DI is uncommon and most cases can be linked to lesions or injury to the hypothalamus after pituitary surgery, traumatic brain injury, aneurysmal subarachnoid hemorrhage, and brain death, as well as with tumours, granulomatous infiltration or autoimmune disease. (Table 5) Damage to the neurohypophyseal stalk during neurosurgery or by trauma can result in a classic triphasic response.81 This syndrome that reflects inhibited release of AVP due to hypothalamic dysfunction typically manifests as early (<24 h) postoperative polyuria lasting 3–5 days. The second phase is characterized by release of stored AVP from the posterior pituitary, often resulting in hyponatremia. Finally, the third phase can again be characterized by central DI, due to potentially permanent hypothalamic dysfunction that usually occurs in 5–10 days once stored AVP is completely depleted from the posterior pituitary.
Nephrogenic DI refers to polyuria and a urinary concentrating defect resulting from renal resistance to the antidiuretic effects of AVP. There are hereditary forms of nephrogenic DI that are usually encountered in children and less commonly in critically ill patients. Hereditary nephrogenic DI can result from either gene mutations to the AVP-2 receptor or to the AQP-2 water channels. Acquired nephrogenic DI is typically related to either AVP resistance at the site of action in the distal tubule or collecting ducts or interference in the medullary countercurrent mechanism, causing impaired renal concentrating capacity. Lithium toxicity and metabolic abnormalities, particularly hypokalemia and hypercalcemia, are the most common causes of acquired nephrogenic DI, but numerous other etiologies have been implicated82 (Table 6). Long-term lithium therapy can lead to polyuria and impaired renal concentrating defects in an estimated 20–30% of patients and DI in 10–12% of patients, due to the down-regulation of AVP-2 receptors and/or reduced expression of AQP-2 channels.83,84 Hypercalcemia (serum [Ca+] > 2.75 mmol l−1) can impair maximal urine concentrating capacity by causing a reversible defect in Na+ and Cl− reabsorption in the ascending loop of Henle and by decreased expression and/or function of the AQP-2 channels. Similar to lithium and hypercalcemia, persistent hypokalemia (serum [K+] < 2.5 mmol l−1) can decrease the renal responsiveness to AVP through reduced AQP-2 expression and/or function and diminished thick ascending loop reabsorption of Na+ and Cl−.
Other renal hypotonic fluid losses
There are several additional renal causes of hypotonic fluid losses.
Osmotic diuresis is caused by an excess of urinary solute, typically non-reabsorbable, that induces polyuria and hypotonic fluid loss. Osmotic diuresis can result from hyperglycemia (i.e., diabetic ketoacidosis), use of mannitol, increased serum urea concentration, or administration of other hypertonic therapies.
The use of diuretics (i.e., loop or thiazide diuretics) is also common in critically ill patients and can contribute to hypotonic urinary fluid losses.
The relief of complete post-renal urinary obstruction can initially be associated with a large diuresis. While much of this diuresis may be appropriate, there may also be a mild urinary concentrating defect due to down-regulation of AQP-2 channels that can predispose to significant hypotonic fluid loss.
Non-renal hypotonic fluid losses
Insensible fluid losses from the skin (i.e., sweat) and from the respiratory tract (i.e., evaporation) are generally hypotonic to serum; hence, if losses are not replaced, hypernatremia will ensue in circumstances of increased insensible fluid loss, such a fever, diaphoresis or tachypnea. Critically ill patients with burns or postoperative patients with open abdominal or other surgical wounds may be at risk for greater insensible fluid loss and need to be monitored accordingly.
Fluid losses from the gastrointestinal tract are also generally hypotonic to serum, and, consequently, will lead to hypernatremia if not replaced. These losses can occur from vomiting, nasogastric drainage, enterocutanous fistulas, or diarrhea. The use of osmotic cathartic agents (i.e., lactulose) or various oral medication suspensions (i.e., sorbitol) can also lead to hypotonic fluid losses.
Water shift into cells
Transient hypernatremia can be induced by intense exercise or by prolonged convulsive seizure activity.85,86 This phenomenon typically occurs in the context of marked lactic acidosis and can transiently raise serum [Na+] by 10–15 mmol l−1. The breakdown of glycogen into osmotically more active solutes acutely raises intracellular osmolality and, as a consequence, induces a shift of hypotonic fluid from the extracellular to the intracellular compartment. The serum [Na+] generally returns to normal in ten to 15 min and is not associated with any apparent sequelae.
Sodium overload
Acute and often severe hypernatremia can be induced by administering hypertonic solutions containing sodium or by ingesting a massive amount of salt.
In critically ill patients, the administration of sodium bicarbonate for a range of conditions, such as metabolic acidosis, tricyclic antidepressant overdose, and rhabdomyolysis, can potentially lead to hypervolemic hypernatremia. Similarly, hypernatremia can occur with hypertonic saline treatment, as may be used to manage intracranial hypertension in traumatic brain injury.
Enteral nutrition with hyperosmolar or high protein feeds accompanied by insufficient free water may lead to hypernatremia, particularly in patients receiving chronic nutritional support.
Numerous reports of severe hypernatremia after surgery with hypertonic saline irrigation for hydatid cysts (echinococcus granulosus) have been reported.87–89 Iatrogenic hospital-acquired hypernatremia has also been reported with use of hypertonic saline in gastric lavage and hypertonic saline-soaked wound packs for gas gangrene.90,91
There are also several reports of accidental or non-accidental acute salt poisoning and extreme hypernatremia, due to massive ingestion of table salt or use of salt or hypertonic saline as an emetic.92,93
Principles of management of hypernatremia
In the approach to the clinical management of the patient with hypernatremia, there are three essential questions that should be asked:
-
1.
What is the underlying diagnosis of hypernatremia, and, if known, is there an etiology-specific treatment?
-
2.
What rate of correction of serum [N+] is considered safe, given the clinical context?
-
3.
What is the volume of free water that is needed to raise the serum [N+] and correct the deficit?
Underlying diagnosis and etiology-specific treatment
The initial step in managing the patient with hypernatremia is confirming the diagnosis and instituting and/or discontinuing cause-specific predisposing factors. For example, this may include preventing further gastrointestinal or insensible fluid losses (i.e., treating fever), treating hyperglycemia and glucosuria, and adjusting enteric-parenteral feeding solutions.
Diabetes insipidus causes hypernatremia by inducing polyuria and renal free water loss. Management should be directed at the underlying precipitating factor(s), when possible; at strategies to reduce urine output, and at replacement of previous and ongoing fluid losses. In central DI, where the primary defect is AVP deficiency, patients with normal mental status and access to free water often have mild hypernatremia, due to the compensatory stimulation of thirst. However, hypernatremia can develop rapidly in those with altered mental status, impaired thirst mechanisms, and/or reduced access to free water. In general, the polyuria associated with DI can usually be controlled by hormone replacement with AVP analogues that have potent anti-diuretic properties, such as desmopressin (dDAVP) (intranasal 5–20 μg once or twice per day; oral 0.05–0.8 mg in divided doses per day; and subcutaneous 1 μg every 12 h).94,95 Desmopressin use is generally considered safe; however, it can be associated with an increased risk for development of hyponatremia, due to impaired free water excretion from non-suppressible AVP activity after administration, particularly if free water intake or administration is continued.95,96 Additional medications, either alone or in combination with desmopressin, may occasionally be needed to control polyuria for the chronic management of central DI. These can be used to increase AVP release (clofibrate 500 mg every 6 h), to enhance renal response to AVP or desmopressin (chlorpropamide 125 mg once or twice per day; carbamazepine 100–300 mg twice daily), or to act to reduce urine output independent of AVP (hydrochlorothiazide 25–50 mg once or twice per day; selected therapy with non-steroidal anti-inflammatory drugs). In nephrogenic DI, where the primary defect results from partial or complete renal resistance to AVP after correction of potential precipitation factors, the management is generally aimed at reducing polyuria by a combination of low sodium-low protein diet (reduced solute excretion), thiazide diuretics (mild volume depletion), and/or selective use of non-steroidal anti-inflammatory drugs (altered renal prostaglandin synthesis). For those patients with partial resistance, supplemental dDAVP may increase the renal response to AVP.97
In patients with hypothalamic lesions, whereby the stimulus for thirst is impaired, management can be challenging and may resort to forced water intake to maintain normal or near normal serum [Na+]. Therapy for essential hypernatremia, whereby osmostat has been reset, remains uncertain; however, this condition is more often chronic, mild, and asymptomatic.
Primary sodium overload can lead to acute and severe hypernatremia, usually after administration of hypertonic sodium-containing solutions (i.e., hypertonic saline, sodium bicarbonate) or massive salt ingestion. In patients with preserved kidney function, the excess sodium is generally excreted rapidly in the urine. Serum [Na+] correction can be further augmented by the addition of a loop diuretic to promote diuresis along with replacement of urine fluid losses with electrolyte-free water. Management can be more challenging for patients with acute kidney injury and/or with pre-existing chronic kidney disease where sodium elimination is impaired. These patients should be considered for early initiation of renal replacement therapy, particularly if symptomatic.
Rate of correction of serum [Na+]
There is a scarcity of clinical data on defining the safest rate to correct the water deficit (and raise serum [Na+]) in hypernatremia. Likely, acute hypernatremia that develops within a few hours (i.e., accidental sodium poisoning) can be rapidly corrected (1 mmol l−1 per hour), due to inadequate time for the brain to adapt to cerebral dehydration.98 However, hypernatremia that lasts longer than 1–2 days leads to cerebral acclimatization, whereby intracellular volume is restored via water movement from cerebral spinal fluid and by intracellular solute uptake. After this phase, overly rapid correction of serum [Na+] may predispose the patient to cerebral edema and serious neurologic sequelae (analogous to rapid onset hyponatremia) characterized by seizures, coma, and death. Therefore, current recommendations for patients with hypernatremia lasting for a long or unknown duration are for a rate of correction of ≤0.5 mmol l−1 per hour, with a maximum correction of 10–12 mmol l−1 per 24-h period to a target initial serum [Na+] 145 mmol l−1.56
Estimating the free water deficit
Hypernatremia is most commonly caused by an excess loss of water relative to sodium. This usually occurs due to unreplaced losses from the gastrointestinal, genitor-urinary, and/or respiratory systems. In hypernatremic patients, an estimate of the free water deficit can be calculated from the formula56:
TBW refers to estimated current total body water. Normal total body water for men and women is approximately 0.6 and 0.5 times the lean body weight, respectively. However, this estimate may differ in elderly patients and in those with significant dehydration; thus, a more conservative estimate may be necessary (i.e., 0.5 and 0.4 times the lean body weight for men and women, respectively).56
It is important to recognize that this formula only provides an estimate of the free water deficit or the positive water balance that is needed to restore serum [N+] to 140 mmol l−1. This estimate for water replacement does not account for ongoing water losses (i.e., insensible, urine output, gastrointestinal tract). Similarly, this formula does not account for iso-osmotic fluid losses (i.e., osmotic diuresis or diarrhea) that may contribute to ECV depletion. As a consequence, when determining the amount and rate of free water replacement, these ongoing losses must be considered in addition to the pre-existing deficit.
Conclusions
Critically ill patients and those undergoing major surgical interventions frequently have disorders of sodium and water balance that are often iatrogenic. These disorders are generally categorized as either hypo-osmolar or hyper-osmolar, depending on the balance (i.e., excess or deficit) of total body water relative to total body sodium content. More classically recognized as hyponatremia and hypernatremia, these disorders may represent a surrogate for increased neurohormonal activation, organ dysfunction, and worsening severity of illness or progression of underlying chronic disease. Hyponatremia and hypernatremia both require timely recognition and appropriate intervention, in order to prevent an increase in morbidity and mortality that may accompany these disorders.
References
Bennani SL, Abouqal R, Zeggwagh AA, et al. Incidence, causes and prognostic factors of hyponatremia in intensive care. Rev Med Interne 2003; 24: 224–9.
Adler SM, Verbalis JG. Disorders of body water homeostasis in critical illness. Endocrinol Metab Clin North Am 2006; 35: 873–94, xi.
Boscoe A, Paramore C, Verbalis JG. Cost of illness of hyponatremia in the United States. Cost Eff Resour Alloc 2006; 31: 10.
Anderson RJ, Chung HM, Kluge R, Schrier RW. Hyponatremia: a prospective analysis of its epidemiology and the pathogenetic role of vasopressin. Ann Intern Med 1985; 102: 164–8.
Chung HM, Kluge R, Schrier RW, Anderson RJ. Postoperative hyponatremia. A prospective study. Arch Intern Med 1986; 146: 333–6.
DeVita MV, Gardenswartz MH, Konecky A, Zabetakis PM. Incidence and etiology of hyponatremia in an intensive care unit. Clin Nephrol 1990; 34: 163–6.
Beukhof CM, Hoorn EJ, Lindemans J, Zietse R. Novel risk factors for hospital-acquired hyponatremia: a matched case-control study. Clin Endocrinol 2007; 66: 367–72.
Gill G, Huda B, Boyd A, et al. Characteristics and mortality of severe hyponatremia—a hospital-based study. Clin Endocrinol (Oxf) 2006; 65: 246–9.
Hoorn EJ, Lindemans J, Zietse R. Development of severe hyponatraemia in hospitalized patients: treatment-related risk factors and inadequate management. Nephrol Dial Transplant 2006; 21: 70–6.
Klein L, O’Connor CM, Leimberger CM, et al. Lower serum sodium is associated with increased short-term mortality in hospitalized patients with worsening heart failure: results from the Outcomes of a Prospective Trial of Intravenous Milrinone for Exacerbations of Chronic Heart Failure (OPTIME-CHF) study. Circulation 2005; 111: 2454–60.
Goldberg A, Hammerman H, Petcherski S, et al. Hyponatremia and long-term mortality in survivors of acute ST-elevation myocardial infarction. Arch Intern Med 2006; 166: 781–6.
Heuman DM, Abou-Assi SG, Habib A, et al. Persistent ascites and low serum sodium identify patients with cirrhosis and low MELD scores who are at high risk for early death. Hepatology 2004; 40: 802–10.
Biggins SW, Rodriguez HJ, Bacchetti P, Bass NM, Roberts JP, Terrault NA. Serum sodium predicts mortality in patients listed for liver transplantation. Hepatology 2005; 41: 32–9.
Ellis SJ. Severe hyponatraemia: complications and treatment. QJM 1995; 88: 905–9.
Almond CS, Shin AY, Fortescue EB, et al. Hyponatremia among runners in the Boston Marathon. N Engl J Med 2005; 352: 1550–6.
Palmer BF. Hyponatraemia in a neurosurgical patient: syndrome of inappropriate antidiuretic hormone secretion versus cerebral salt wasting. Nephrol Dial Transplant 2000; 15: 262–8.
Palmer BF. Hyponatremia in patients with central nervous system disease: SIADH versus CSW. Trends Endocrinol Metab 2003; 14: 182–7.
Schrier RW. Body water homeostasis: clinical disorders of urinary dilution and concentration. J Am Soc Nephrol 2006; 17: 1820–32.
Hanna FW, Scanlon MF. Hyponatraemia, hypothyroidism, and role of arginine-vasopressin. Lancet 1997; 350: 755–6.
Davison JM, Shiells EA, Philips PR, Lindheimer MD. Influence of humoral and volume factors on altered osmoregulation of normal human pregnancy. Am J Physiol 1990; 258(4 Pt 2): F900–7.
Oren RM. Hyponatremia in congestive heart failure. Am J Cardiol 2005; 95: 2B–7B.
Papadakis MA, Fraser CL, Arieff AI. Hyponatraemia in patients with cirrhosis. Q J Med 1990; 76: 675–88.
Gillum DM, Linas SL. Water intoxication in a psychotic patient with normal renal water excretion. Am J Med 1984; 77: 773–4.
Stuart CA, Neelon FA, Lebovitz HE. Disordered control of thirst in hypothalamic-pituitary sarcoidosis. N Engl J Med 1980; 303: 1078–82.
Klonoff DC, Jurow AH. Acute water intoxication as a complication of urine drug testing in the workplace. JAMA 1991; 265: 84–5.
Brvar M, Kozelj G, Osredkar J, Mozina M, Gricar M, Bunc M. Polydipsia as another mechanism of hyponatremia after ‘ecstasy’ (3,4 methyldioxymethamphetamine) ingestion. Eur J Emerg Med 2004; 11: 302–4.
Hilden T, Svendsen TL. Electrolyte disturbances in beer drinkers. A specific “hypo-osmolality syndrome”. Lancet 1975; 2: 245–6.
Thaler SM, Teitelbaum I, Berl T. “Beer potomania” in non-beer drinkers: effect of low dietary solute intake. Am J Kidney Dis 1998; 31: 1028–31.
Palevsky PM, Rendulic D, Diven WF. Maltose-induced hyponatremia. Ann Intern Med 1993; 118: 526–8.
Hillier TA, Abbott RD, Berrett EJ. Hyponatremia: evaluating the correction factor for hyperglycemia. Am J Med 1999; 106: 399–403.
Arieff AI. Hyponatremia, convulsions, respiratory arrest, and permanent brain damage after elective surgery in healthy women. N Engl J Med 1986; 314: 1529–35.
Arieff AI, Ayus JC, Fraser CL. Hyponatraemia and death or permanent brain damage in healthy children. BMJ 1992; 304: 1218–22.
Ayus JC, Wheeler JM, Arieff AI. Postoperative hyponatremic encephalopathy in menstruant women. Ann Intern Med 1992; 117: 891–7.
Ferreira da Cunha D, Pontes Monteiro J, Modesto dos Santos V, Araujo Oliveira F, Freire de Carvalho da Cunha S. Hyponatremia in acute-phase response syndrome patients in general surgical wards. Am J Nephrol 2000; 20: 37–41.
Madiba TE, Haffejee AA, Mokoena TR. Hyponatraemia—a prospective analysis of surgical patients. S Afr J Surg 1998; 36: 78–81.
McPherson E, Dunsmuir RA. Hyponatraemia in hip fracture patients. Scott Med J 2002; 47: 115–6.
Tambe AA, Hill R, Livesley PJ. Post-operative hyponatraemia in orthopaedic injury. Injury 2003; 34: 253–5.
Pasini A, Belloni C. Intraoperative complications of 697 consecutive operative hysteroscopies (Italian). Minerva Ginecol 2001; 53: 13–20.
Gardner LB, Preston RA. University of Miami Division of Clinical Pharmacology Therapeutic Rounds: the water-intolerant patient and perioperative hyponatremia. Am J Ther 2000; 7: 23–30.
Bhananker SM, Paek R, Vavilala MS. Water intoxication and symptomatic hyponatremia after outpatient surgery. Anesth Analg 2004; 98: 1294–6.
Amede FJ, James KA, Michelis MF, Gleim GW. Changes in serum sodium, sodium balance, water balance, and plasma hormone levels as the result of pelvic surgery in women. Int Urol Nephrol 2002; 34: 545–50.
Guglielminotti J, Pernet P, Maury E, et al. Osmolar gap hyponatremia in critically ill patients: evidence for the sick cell syndrome? Crit Care Med 2002; 30: 1051–5.
Guglielminotti J, Tao S, Maury E, Fierobe L, Mantz J, Desmonts JM. Hyponatremia after hip arthroplasty may be related to a translocational rather than to a dilutional mechanism. Crit Care Med 2003; 31: 442–8.
Gill GV, Osypiw JC, Shearer E, English PJ, Watson ID. Critical illness with hyponatraemia and impaired cell membrane integrity—the “sick cell syndrome” revisited. Clin Biochem 2005; 38: 1045–8.
Benito Ruiz J, Baena Montilla P, Navarro Monzonis A. Sick cell syndrome in a burned patient. Burns 1990; 16: 309–12.
Rhymer JC, Bell TJ, Perry KC, Ward JP. Hyponatraemia following transurethral resection of the prostate. Br J Urol 1985; 57: 450–2.
Gonzales R, Brensilver JM, Rovinsky JJ. Posthysteroscopic hyponatremia. Am J Kidney Dis 1994; 23: 735–8.
Michielsen DP, Debacker T, De Boe V, et al. Bipolar transurethral resection in saline—an alternative surgical treatment for bladder outlet obstruction? J Urol 2007; 178: 2035–9; discussion 2039.
Finley DS, Beck S, Szabo RJ. Bipolar saline TURP for large prostate glands. ScientificWorldJournal 2007; 7: 1558–62.
Issa MM, Young MR, Bullock AR, Bouet R, Petros JA. Dilutional hyponatremia of TURP syndrome: a historical event in the 21st century. Urology 2004; 64: 298–301.
Ho H, Yip SK, Cheng CW, Foo KT. Bipolar transurethral resection of prostate in saline: preliminary report on clinical efficacy and safety at 1 year. J Endourol 2006; 20: 244–6; discussion 246–7.
Ho HS, Yip SK, Lim KB, Fook S, Foo KT, Cheng CW. A prospective randomized study comparing monopolar and bipolar transurethral resection of prostate using transurethral resection in saline (TURIS) system. Eur Urol 2007; 52: 517–22.
Verbalis JG, Goldsmith SR, Greenberg A, Schrier RW, Sterns RH. Hyponatremia treatment guidelines 2007: expert panel recommendations. Am J Med 2007; 120(11 Suppl 1): S1–21.
Sterns RH, Cappuccio JD, Silver SM, Cohen EP. Neurologic sequelae after treatment of severe hyponatremia: a multicenter perspective. J Am Soc Nephrol 1994; 4: 1522–30.
Soupart A, Decaux G. Therapeutic recommendations for management of severe hyponatremia: current concepts on pathogenesis and prevention of neurologic complications. Clin Nephrol 1996; 46: 149–69.
Adrogue HJ, Madias NE. Hypernatremia. N Engl J Med 2000; 342: 1493–9.
Ghali JK, Koren MJ, Taylor JR, et al. Efficacy and safety of oral conivaptan: a V1A/V2 vasopressin receptor antagonist, assessed in a randomized, placebo-controlled trial in patients with euvolemic or hypervolemic hyponatremia. J Clin Endocrinol Metab 2006; 91: 2145–52.
Gheorghiade M, Niazi I, Ouyang J, et al. Vasopressin V2-receptor blockade with tolvaptan in patients with chronic heart failure: results from a double-blind, randomized trial. Circulation 2003; 107: 2690–6.
Konstam MA, Gheorghiade M, Burnett JC Jr, et al. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA 2007; 297: 1319–31.
Schrier RW, Gross P, Gheorghiade M, et al. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med 2006; 355: 2099–112.
Zeltser D, Rosansky S, van Rensburg H, Verbalis JG, Smith N; Conivaptan Study Group. Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia. Am J Nephrol 2007; 27: 447–57.
Josiassen RC, Goldman M, Jessani M, et al. Double-blind, placebo-controlled, multicenter trial of a vasopressin V(2)-receptor antagonist in patients with schizophrenia and hyponatremia. Biol Psychiatry 2008.
Tanneau RS, Henry A, Rouhart F, et al. High incidence of neurologic complications following rapid correction of severe hyponatremia in polydipsic patients. J Clin Psychiatry 1994; 55: 349–54.
Snyder NA, Feigal DW, Arieff AI. Hypernatremia in elderly patients. A heterogenous, morbid, and iatrogenic entity. Ann Intern Med 1987; 107: 309–19.
Palevsky PM, Bhagrath R, Greenberg A. Hypernatremia in hospitalized patients. Ann Intern Med 1996; 124: 197–203.
Polderman KH, Schreuder WO, Strack van Schijndel RJ, Thijs LG. Hypernatremia in the intensive care unit: an indicator of quality of care? Crit Care Med 1999; 27: 1105–8.
Molaschi M, Ponzetto M, Massaia M, Villa L, Scarafiotti C, Ferrario E. Hypernatremic dehydration in the elderly on admission to hospital. J Nutr Health Aging 1997; 1: 156–60.
Hoorn EJ, Betjes MG, Weigel J, Zietse R. Hypernatraemia in critically ill patients: too little water and too much salt. Nephrol Dial Transplant 2008; 23: 1562–8.
Lindner G, Funk GC, Schwarz C, et al. Hypernatremia in the critically ill is an independent risk factor for mortality. Am J Kidney Dis 2007; 50: 952–7.
Moder KG, Hurley DL. Fatal hypernatremia from exogenous salt intake: report of a case and review of the literature. Mayo Clin Proc 1990; 65: 1587–94.
Holley AD, Green S, Davoren P. Extreme hypernatraemia: a case report and brief review. Crit Care Resusc 2007; 9: 55–8.
Park YJ, Kim YC, Kim MO, Ruy JH, Han SW, Kim HJ. Successful treatment in the patient with serum sodium level greater than 200 mEq/L. J Korean Med Sci 2000; 15: 701–3.
Portel L, Hilbert G, Gruson D, Gbikpi-Benissan G, Cardinaud JP. Survival with extreme hypernatremia at 209 mmol/l. Intensive Care Med 1998; 24: 197–8.
Borrego Dominguez RR, Imaz Roncero A, Lopez-Herce Cid J, Serina Ramirez C. Severe hypernatremia: survival without neurologic sequelae (Spanish). An Pediatr (Barc) 2003; 58: 376–80.
Gomez-Daspet J, Elko L, Grebenev D, Vesely DL. Survival with serum sodium level of 180 mEq/L: permanent disorientation to place and time. Am J Med Sci 2002; 324: 321–5.
van der Helm-van Mil AH, van Vugt JP, Lammers GJ, Harinck HI. Hypernatremia from a hunger strike as a cause of osmotic myelinolysis. Neurology 2005; 64: 574–5.
Reynolds RM, Padfield PL, Seckl JR. Disorders of sodium balance. BMJ 2006; 332: 702–5.
Phillips PA, Rolls BJ, Ledingham JG, et al. Reduced thirst after water deprivation in healthy elderly men. N Engl J Med 1984; 311: 753–9.
Phillips PA, Bretherton M, Johnston CI, Gray L. Reduced osmotic thirst in healthy elderly men. Am J Physiol 1991; 261(1 Pt 2): R166–71.
Robertson GL. Abnormalities of thirst regulation. Kidney Int 1984; 25: 460–9.
Ultmann MC, Hoffman GE, Nelson PB, Robinson AG. Transient hyponatremia after damage to the neurohypophyseal tracts. Neuroendocrinology 1992; 56: 803–11.
Garofeanu CG, Weir M, Rosas-Arellano MP, Henson G, Garg AX, Clark WF. Causes of reversible nephrogenic diabetes insipidus: a systematic review. Am J Kidney Dis 2005; 45: 626–37.
Bendz H, Aurell M. Drug-induced diabetes insipidus: incidence, prevention and management. Drug Saf 1999; 21: 449–56.
Bendz H, Aurell M, Balldin J, Mathe AA, Sjodin I. Kidney damage in long-term lithium patients: a cross-sectional study of patients with 15 years or more on lithium. Nephrol Dial Transplant 1994; 9: 1250–4.
Nose H, Takamata A, Mack GW, et al. Water and electrolyte balance in the vascular space during graded exercise in humans. J Appl Physiol 1991; 70: 2757–62.
Felig P, Johnson C, Levitt M, Cunningham J, Keefe F, Boglioli B. Hypernatremia induced by maximal exercise. JAMA 1982; 248: 1209–11.
Albi A, Baudin F, Matmar M, Archambeau D, Ozier Y. Severe hypernatremia after hypertonic saline irrigation of hydatid cysts. Anesth Analg 2002; 95: 1806–8.
Gage TP, Vivian G. Hypernatremia after hypertonic saline irrigation of an hepatic hydatid cyst. Ann Intern Med 1984; 101: 405.
Wanninayake HM, Brough W, Bullock N, Calne RY, Farman JV. Hypernatraemia after treatment of hydatid. Br Med J (Clin Res Ed) 1982; 284: 1302–3.
Mofredj A, Rakotondreantoanina JR, Farouj N. Severe hypernatremia secondary to gastric lavage (French). Ann Fr Anesth Reanim 2000; 19: 219–20.
Thorp JM, Mackenzie I, Simpson E. Gross hypernatraemia associated with the use of antiseptic surgical packs. Anaesthesia 1987; 42: 750–3.
Casavant MJ, Fitch JA. Fatal hypernatremia from saltwater used as an emetic. J Toxicol Clin Toxicol 2003; 41: 861–3.
Ward DJ. Danger of saline emetics. Br Med J 1977; 2: 459.
Richardson DW, Robinson AG. Desmopressin. Ann Intern Med 1985; 103: 228–39.
Vande Walle J, Stockner M, Raes A, Norgaard JP. Desmopressin 30 years in clinical use: a safety review. Curr Drug Saf 2007; 2: 232–8.
Kelleher HB, Henderson SO. Severe hyponatremia due to desmopressin. J Emerg Med 2006; 30: 45–7.
Stasior DS, Kikeri D, Duel B, Seifter JL. Nephrogenic diabetes insipidus responsive to indomethacin plus dDAVP. N Engl J Med 1991; 324: 850–1.
Lien YH, Shapiro JI, Chan L. Effects of hypernatremia on organic brain osmoles. J Clin Invest 1990; 85: 1427–35.
Conflicts of interest
None declared.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bagshaw, S.M., Townsend, D.R. & McDermid, R.C. Disorders of sodium and water balance in hospitalized patients. Can J Anesth/J Can Anesth 56, 151–167 (2009). https://doi.org/10.1007/s12630-008-9017-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12630-008-9017-2