
Abstract
Purpose of Review
The I-SPY 2 trial is an adaptive clinical trial platform designed to improve outcomes in high-risk breast cancer patients by testing new drugs in the neoadjuvant setting. The intent of this review is to discuss background, study structure, innovation, and outcomes of the I-SPY 2 trial.
Recent Findings
I-SPY 2 evaluates new agents combined with standard therapy with pathologic complete response (pCR) as the primary endpoint. I-SPY-2 uses clinical biomarkers to classify breast cancer into 10 subtypes, with Bayesian adaptive randomization to allow individualized patient assignment to therapy arms to maximize treatment effects. A total of 7 drugs have graduated from I-SPY 2. Multiple new agents are currently in active enrollment in I-SPY 2.
Summary
I-SPY 2 uses an individualized approach in clinical trial design to improve high-risk breast cancer outcomes. The purpose of this review is to encourage further research and innovation in this area and bring more precise treatment options to breast cancer patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Despite the progress in treatment of breast cancer over the years, some patients continue to have a high risk of recurrence and death even when systemic therapy was employed [1, 2]. Additional treatment options are needed to improve the outcome of this subgroup of patients who recur despite receiving recommended adjuvant therapy.
Traditional trials frequently relied upon a relatively standardized battery of preclinical tests followed by clearly defined and generally understood sequential clinical development steps requiring a large numbers of patients with extended follow-up to meet discrete predefined regulatory milestones required for drug approval [3]. In the past, most new cancer drugs were first tested in metastatic disease, followed by randomized phase 3 trials in the adjuvant setting without assigning therapies based on the molecular characteristics of each patient’s disease. Recurrence free survival (RFS) and overall survival (OS) were traditionally used as endpoints. With such an endpoint, thousands of patients were needed and long-term follow-up (10–20 years) was required to gain approval of a promising drug, resultantly a substantial number of drugs ultimately fail this process [4]. Most clinicians knew this approach was imprecise. While the combination of tamoxifen and chemotherapy in estrogen receptor alpha (ERα)–positive breast cancer was superior to tamoxifen alone, it was evident that many patients had exceptional outcomes with tamoxifen alone [5]. Gene expression profiling has proven this point; most patients do not benefit from the addition of chemotherapy to endocrine therapy [6, 7].
Several factors have argued strongly for a need to change the way adjuvant clinical trials are performed. First, it has been increasingly expensive to develop new drugs following the traditional pathway of testing first in the metastatic setting. The estimated average cost per approved new compound is close to $1.5 billion (2013 dollars) [8]. Oncology drugs may have a high clinical failure rate due to the heterogeneity of many cancers. It has always been a challenge to identify specific cancer subtypes responsive to a specific drug and incorporate this into a clinical trial design. The heterogeneity of breast cancer makes it difficult to see how molecular targeted therapies will benefit all subtypes of breast cancer.
Second, the heterogeneity needs to be addressed in an efficient and timely way. There is no clearer example of this issue than the development of trastuzumab in the adjuvant therapy of breast cancer. While the benefit of adding trastuzumab to chemotherapy in human epidermal growth factor receptor 2 (HER2)–over-expressing patients was striking, it took 50 months to accrue 3270 women with 46 months of follow-up to show this benefit was not seen in HER2-low patients [9, 10]. This example demonstrates the critical importance of evaluating and selecting therapies based on tumor biomarkers. Without a strong predictive biomarker, there are real-life consequences for patients as well as opportunity costs in conducting an inefficient trial.
Third, many compounds are potentially promising anticancer agents. Basic research has highlighted the many ways cancer cells maintain sustained proliferation, resistance to apoptosis, and initiate metastases. Many drugs have been created to target these pathways; there are more than can be tested by the traditional pathway if a drug’s benefit may only apply to a minority of patients with breast cancer. It is becoming increasingly evident that the traditional clinical trial and drug approval process appears to be overly cumbersome and costly, and has become a barrier to new treatment to patients especially with the availability of our latest biomarker guidance. In the end, it is cancer patients who suffer from a lack of effective treatment and from the financial burden related to the high cost of cancer treatment. I-SPY 2 fills the need for novel phase 2 trials, which can rapidly evaluate and select promising compounds for phase 3 testing and get the latest treatments to patients in need.
I-SPY 2 TRIAL (Investigation of Serial studies to Predict Your Therapeutic Response with Imaging and Molecular AnaLysis 2, NCT01042379) has the framework of an adaptive phase 2 clinical trial design in the neoadjuvant setting for women with high-risk clinical stage II or III breast cancer. It is designed to evaluate multiple new agents added to standard chemotherapy concurrently with the primary endpoint of pathologic complete response (pCR). The goal of the I-SPY 2 trial is to target rapid, focused, and individualized clinical development of promising agents or agent combinations based upon breast cancer biomarker subtypes. It was launched in 2010 and is, by far, the longest running adaptive platform trial.
I-SPY 2 Trial Structure
Initially led by the Foundation for the National Institutes of Health (FNIH), the Biomarkers Consortium (a unique public-private partnership including the U.S. Food and Drug Administration (FDA), the National Institutes of Health (NIH), and major pharmaceutical companies) designed the I-SPY 2 trial to explore an innovative way to rapidly screen new agents or combinations and match the therapy to specific markers in breast cancer patients at high risk of early recurrence. It is currently administratively supported by Quantum Leap Healthcare Collaborative (QLHC) and continues to enroll patients at 16 clinical study sites around the USA.
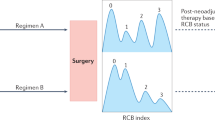
I-SPY 2 is an ongoing, multicenter, open-label, adaptive phase 2 platform trial with multiple experimental groups to evaluate new agents combined with standard neoadjuvant therapy (Fig. 1).

I-SPY 2 treatment schema adapted (from https://www.ispytrials.org/i-spy-platform/i-spy2, Accessed on June 20, 2019. © 2019 Quantum Leap Healthcare Collaborative)
Eligible women are over 18 years of age with good performance status (ECOG performance status 0–1), have stage II or stage III breast cancer with a tumor 2.5 cm or larger in diameter by imaging or physical examination, and have no prior surgery or systemic therapy. A common control group receiving standard neoadjuvant therapy was used for all experimental groups. The control group receives standard neoadjuvant therapy consisting of weekly intravenous paclitaxel (T) at a dose of 80 mg per m2 of body surface area for 12 cycles, followed by 4 cycles of doxorubicin (A) at a dose of 60 mg per m2 and cyclophosphamide (C) at a dose of 600 mg per m2, administered intravenously every 2 weeks to 3 weeks. Patients who have HER2+ cancer also receives trastuzumab for the first 12 weeks, with a loading dose of 4 mg per kg of body weight in the first cycle, followed by a maintenance dose of 2 mg per kg in cycles 2 through 12 [11••]. Pertuzumab (P) of 840 mg IV (loading dose) for week 1 and 420 mg every 3 weeks (weeks 4, 7, and 10) was added to the standard therapy after receiving accelerated approval from the FDA [12, 13].
In the experimental arms, a new drug is added to standard neoadjuvant regimen and given concurrently with weekly paclitaxel for the first 12 weeks. New drugs must meet specific criteria of safety and efficacy before entering I-SPY 2 to ensure patient safety. Five new drugs may be tested simultaneously, with no more than 120 patients tested for each experimental arm [14]. Multiple biopsies and magnetic resonance imaging (MRI) scans are obtained throughout the trial to assess response. Further, biopsies prior to, during, and after treatment are used to discover new predictive biomarkers.
The primary endpoint is pCR, defined as elimination of invasive cancer in the breast and lymph nodes at the time of surgery. Tumor volume changes on serial MRI are used to assess the likelihood of such a response in each patient during the treatment process at the time of enrollment, then week 3, week 12, and before surgery [14]. Following completion of neoadjuvant chemotherapy, patients undergo surgery including axillary node sampling according to the National Comprehensive Cancer Network and local practice guidelines.
In I-SPY 2, breast cancer is categorized into molecular subtypes on the basis of hormone receptor (HR) status, HER2 status, and risk according to a 70-gene assay (MammaPrint, Agendia) [7, 15]. The trial classifies breast cancer into 10 subtypes based on ER, HER2, and MammaPrint scores. The molecular subtype is used to adaptively randomize patients to control arm or experimental arms.
Bayesian methods of adaptive randomization are applied in I-SPY 2 to achieve a higher probability of efficacy while assigning patients to different arms [16]. Drugs that increase pCR rates within a specific molecular subtype will be preferentially assigned to new patients entering the trial with that particular subtype allowing for a more rapid determination of success. On the other hand, drugs doing poorly within a particular subtype will be less likely assigned to that subtype. Bayesian adaptive randomization is used during the entire process; the likelihood of assignment to a given agent or combination increases as the trial continues and evidence accrues as more efficacious than the control in achieving pCR. Enrollment in the experimental group will be stopped when Bayesian predictive probability of success reaches a prespecified threshold (usually 85%) for any biomarker signature in a confirmatory 300-patient phase 3 trial of neoadjuvant therapy [14]. When that occurs, the agent/combination is deemed as “graduated” from the trial. Enrollment will also be stopped for futility if the probability falls to below 10% for all biomarker signatures; the drug is then dropped from the trial [14]. Depending on the patient accrual rate, new agents can be added to the trial at any time as agents being tested are either graduated or dropped.
The I-SPY 2 trial has strict safety monitoring process to ensure patient safety. Experimental drugs must meet specific criteria relating to safety and efficacy to enter the I-SPY 2 trial [17]. A candidate drug must have been tested and demonstrated safety data in at least one phase 1 clinical study with a combination of taxane (or taxane plus trastuzumab for HER2-positive subjects) [14]. It must have been reviewed and approved by an independent advisory committee and the FDA before it is trial eligible. Additionally, laboratory and adverse event data are collected and monitored in real time and an external independent data safety and monitoring board (DSMB) meets monthly to review toxicity within each experimental agent/regimen of the trial [18]. Patient safety has been of the utmost importance in I-SPY 2 trial design and patient advocates are involved in all aspects of trial design, drug review, safety assessment, and conduct.
Innovation
The I-SPY 2 trial pioneers many aspects in phase 2 trial design. The most important aspects are the use of pCR as an early surrogate primary endpoint to enable rapid evaluation of promising agents/combinations, individualized treatment by Bayesian adaptive randomization to identify benefit from new agents, ability to allow multiple drugs to be tested at the same time, and robust biomarker collection including multiple biopsies and imaging. This foundational data can be used to move forward to confirmatory phase 3 trials.
Early Surrogate Endpoint
Recently, neoadjuvant therapy for breast cancer has gained in popularity as a standard-of-care clinical option. Neoadjuvant therapy can downstage large cancers to improve the breast conservation rate, and it also provides the unique opportunity to assess tumor response to therapy in vivo and helps to guide further treatment. I-SPY 2 is conducted in a neoadjuvant setting. pCR was designated as the primary endpoint with the goal of rapidly evaluating drugs under investigation. This primary endpoint is evident immediately after surgery—within 24 weeks of beginning chemotherapy. In contrast, traditional clinical trials may take years to evaluate the primary endpoints of event-free survival (EFS), distant recurrence-free survival (DRFS), and Overall Survival (OS) [8, 19•]. Using pCR as the surrogate primary endpoint can significantly shorten the evaluation process of promising drugs. In I-SPY 2, the average time for a drug to “graduate” is only about 18 months and is much shorter than the duration of a traditional phase 2 trial [19•].
Although previously reported neoadjuvant trials showed a strong association between long-term outcome and pCR, the strength of relationship between improvements in the early measure (pCR) for specific drug regimens and later clinically relevant endpoints (EFS, DRFS, OS) has been questioned [20, 21]. There have been many debates over the utility of pCR as a surrogate early endpoint. Because of that, the FDA conducted the Collaborative Trials in Neoadjuvant Breast Cancer (CTNeoBC), with pooled patient-level meta-analysis of 11,955 patients in 12 randomized clinical trials to assess the relationship between pCR and EFS and OS. This analysis showed a long-term benefit for patients achieving pCR; the overall EFS hazard ratio was 0.48 for pCR versus non-pCR. The long-term benefit exists for triple-negative (HR = 0.24), HER2-positive (HR = 0.39), and Hormone Receptor positive/HER2-negative (HR = 0.49) diseases [22]. The FDA then issued guidance in 2014, indicating pCR could be used as a surrogate endpoint for accelerated approval coupled with a confirmatory trial in an adjuvant or neoadjuvant setting demonstrating a clinically meaningful and statistically significant improvement in EFS or OS [23]. There remains controversy regarding the improvement in pCR rates attributed to specific drug regimens and the relationship to improved EFS. Modeling of this relationship suggests it varies by subtype, and the ability of one regimen’s superiority over another based on pCR must be taken into consideration when designing a confirmatory study [24]. However, it is evident for an individual patient, in which obtaining a pCR has substantial prognostic implications [22].
Results from I-SPY 1 trial (using an AC-paclitaxel regimen) showed HR-negative/HER2-positive patients had highest pCR (54%), HR-positive/HER2-negative patients had lowest pCR (9%), and achieving pCR predicted favorable RFS [25]. One of the predefined goals of I-SPY 2 was also to evaluate the primary endpoint of pCR as a surrogate for the clinically meaningful secondary endpoints such as EFS and DRFS. The long-term I-SPY 2 efficacy investigation demonstrated achieving pCR was a very strong surrogate endpoint for EFS and DRFS regardless of breast cancer subtype and treatment regimen. The result was presented at San Antonio Breast Cancer Symposium in December 2017 [26•].
Adaptive Multigroup Trial Design
Another innovation in the I-SPY 2 trial is adaptive multigroup trial design. The goal of adaptive design is for investigators to learn as they go and not pursue treatments proven ineffective. Multigroup trial design has the potential to evaluate several promising agents simultaneously and is more efficient than traditionally designed clinical trials. Although the efficacy of multigroup early phase trial has been recognized in the past, I-SPY 2 provides a standing platform to test multiple new agents in the neoadjuvant setting [27]. Importantly, when an agent leaves the trial, it does not signify the end of the study. Instead, new agents will enter the trial in a seamless fashion.
Breast cancer is a heterogeneous disease, and different biomarker subtypes of breast cancer respond differently to a therapy and thus can pose great difficulty in targeted drug development. Traditional phase 2 trials test one investigational agent at a time in all subtypes of breast cancer. The treatment effect can be diluted by the heterogeneity of the disease, so more patients need to be tested before an effect can be observed for any particular agent. As mentioned above, I-SPY 2 uses 10 biomarker signatures to distinguish breast cancer into 10 subtypes. Bayesian adaptive randomization is used to increase the likelihood of assignment of a given therapy when evidence shows the therapy is more effective than control in certain subtypes of high-risk breast cancer in achieving pCR. The system learns and adapts as the trial accrues patients. Assigning the therapy to more specific targeted molecular subtypes of breast cancer may increase the chances of good response [28]. I-SPY 2 has also shown some biomarkers, currently not utilized in standard clinical practice, may further demonstrate efficacy of specific drug regimens [29•]. Meanwhile, the adaptive trial design also decreases the likelihood of exposing a patient to unnecessary toxicity by avoiding treatments for which meaningful benefits are unlikely. This highly individualized approach is the future of early-phase clinical trial design. As cancer treatment is becoming more specifically aimed at different targets, the traditional trial designs will be insufficient to match patients to effective drugs and thus preventing potential patient treatment benefits, wasting resources, time, and exposing patients to drug toxicity without a chance of benefit.
Example of Drugs Graduated from I-SPY 2
Neratinib was one of the agents selected and entered the I-SPY 2 trial in 2010. Neratinib is a dual epidermal growth factor receptor (EGFR) and HER2 irreversible inhibitor. These targets, especially EGFR, are implicated in hormone-resistant ER-positive breast cancer and triple-negative breast cancer (TNBC) [30, 31]. Therefore, neratinib was open to all patients entering I-SPY 2 and a total of 127 participants were enrolled and randomly assigned to receive neratinib; 115 patients were evaluated as 12 withdrew from the trial and 84 patients were randomly assigned to the control group (paclitaxel and trastuzumab). Using the Bayesian model, neratinib reached the prespecified threshold of efficacy in the HER2-positive, HR-negative subtype. Among this subtype, the Bayesian-estimated rate of pCR was 56% (95% probability interval (PI), 37% to 73%) in the neratinib group, compared to 33% (95% PI, 11% to 54%) in the control group [11••]. The analysis resulted in the probability that neratinib was 95% superior to standard therapy, and the probability of neratinib to be a success in a phase 3 clinical trial involving 300 patients was 79% [11••]. Neratinib thus graduated from the I-SPY 2 trial. As neratinib showed very little activity in patients with HER2-negative, HR-positive cancer or with HER2-negative, HR-negative cancer, the adaptive randomization algorithm stopped assigning patients with these subtypes to receive neratinib during the course of trial; thus, a larger percentage of patients with HER2-positive cancer were enrolled in the neratinib group (57%) compared to the control group (28%) [11••]. In July 2017, the FDA approved neratinib for adjuvant therapy in early-stage HER2-positive breast cancer based on the ExterNET trial for patients with residual disease after HER2-based neoadjuvant therapy (NCT00878709) [32].
Veliparib-carboplatin was another drug combination graduated from I-SPY 2. From May 2010 through July 2012, a total of 75 patients were randomly assigned to veliparib-carboplatin-paclitaxel arm (VC), and 44 patients were concurrently randomized into a control group (paclitaxel only); VC regimen was evaluated only for HER2-negative tumors as safety data for this combination with trastuzumab was not available. The benefit of VC was concentrated in triple-negative biomarker subtype, with the estimated pCR rate of 51% (95% PI, 36% to 66%) in the VC group versus 26% (95% PI, 9% to 43%) in the control; in this biomarker subtype, the probability of VC which was superior to control was 99%, and its probability of success in a phase 3 trial including 300 patients was 88% [33•].
These results were validated in the BrighTNess trial [34•]. In this three-arm trial, paclitaxel was compared to either paclitaxel-carboplatin or paclitaxel-carboplatin-veliparib in the neoadjuvant treatment of TNBC. This study enrolled 634 patients over 2 years and showed pCR rates were 58% and 53% for paclitaxel-carboplatin and paclitaxel-carboplatin-veliparib arms, respectively. Both were superior to paclitaxel; pCR rates were similar to those seen in I-SPY 2. In this study, carboplatin was clearly the drug associated with improved pCR, not the veliparib. While this was disappointing regarding the development of a PARP inhibitor in the adjuvant therapy of breast cancer, this study highlights the value of testing a regimen in I-SPY 2. I-SPY 2 data showed the combination could be tested in the appropriate subgroup of breast cancer and essentially identical results were obtained. Further, analysis of I-SPY 2 specimens refined additional biomarkers that could be useful in identifying patients who benefit from the addition of carboplatin, veliparib, or both drugs [29, 35].
The survival benefit of pertuzumab (P) was initially established in the metastatic setting [36]. Pertuzumab’s ability to improve pCR when combined with paclitaxel (T) and Herceptin (H) (THP) was tested in I-SPY 2 trial compared to standard therapy (TH). After accrual of total 75 patients, 44 in THP arm and 31 in TH arm, THP regimen met the Bayesian predictive probability and graduated in three signatures: all HER2-positive, HER2-positive/HR-positive, and HER2-positive/HR-negative [37]. The combination of trastuzumab emtansine (T-DM1) with pertuzumab, but without paclitaxel, in neoadjuvant setting was also tested in I-SPY 2, the control group received standard TH regimen. Fifty-two patients were enrolled into T-DM1+P arm and 31 patients to TH arm. T-DM1+P improved pCR rate and met the predictive probability to graduate from I-SPY 2 in the same three signatures: all HER2-positive, HER2-positive/HR-positive, and HER2-positive/HR-negative [38].
Since its launch in 2010, 17 agents or combinations have entered into the I-SPY 2 trial. Twelve have completed accrual, and 7 drugs have graduated (defined as at least 85% probability of showing statistical significance in a confirmatory phase 3 trial of 300 patients of the specified subtype, randomized 1:1, with pCR as the endpoint) in at least one tumor subtype, with two receiving accelerated approval and one gaining breakthrough designation by the FDA. Table 1 illustrates the agents/combinations entered into the I-SPY 2 trial and their outcome.
Future Considerations
The I-SPY 2 trial is now widely regarded as a model for adaptive platform trials and is a model increasingly adapted for use in other diseases such as glioblastoma and Alzheimer’s disease despite initial concerns raised during its launch [39,40,41]. The success of I-SPY 2 is redefining the standard of modern clinic trials and changing the expectations of breast cancer patients. Yet, the current design of I-SPY 2 is not perfect; improvement and thoughtful modifications are necessary, given the anticipated advances in the standard care of breast cancer.
The current pCR rate in I-SPY 2 neoadjuvant setting is about 35%; that means more than half of stage II or III breast cancer patients still face earlier recurrence and poor outcomes [26•]. On top of identifying new agents to achieve a better response rate, it is worth looking into trial design and creating a way to further individualize the treatment approach and potentially improving the pCR rate by redefining breast cancer subtypes with novel predictive biomarkers and by using current available treatment sequentially. While the recognized phenotypes are used to test individual drug regimens, it is clear Hormone Receptors HER2, and MammaPrint only partially define breast cancer treatment vulnerabilities. As mentioned above, some ER-positive patients benefited from the addition of VC to their treatment regimen based on analysis of DNA repair deficiency biomarkers [29•]. As more antibody-drug conjugates enter clinical practice, it seems likely the expression of the antibody epitope will define a new predictive biomarker independent of Hormone Receptor or HER2. I-SPY 2 can also easily accommodate additional predictive biomarkers of benefit. While pCR is currently used to define benefit, circulating tumor cells and cell-free DNA responses are also being studied to enhance tumor-free biomarker endpoints.
At the same time, I-SPY 2 has an opportunity to minimize or de-escalate therapy. Predefined cycles of treatment may expose patients who respond to initial treatment to unnecessary additional cycles of treatment. Regular tumor monitoring with MRI and biopsy during the I-SPY 2 trial may shorten the treatment duration for patients who show excellent response to initial treatment, thus limiting the exposure to treatment while maintaining the maximum benefit. This may also allow for earlier treatment intervention and modification of treatment for those who do not respond to initial regimen. The goal of I-SPY 2 is not only to identify the new therapies for patients who need them but also to identify patients who can be cured by less treatment, toxicity, and cost; in other words, the goal of I-SPY 2 is to employ a highly individualized approach in managing heterogeneous breast cancer patients.
I-SPY 2 was initially funded by grants, non-profit organizations, and private funds; following FDA-accelerated approval of pertuzumab, I-SPY 2 gained additional support as a business model for new agents where pharmaceutical companies covered the full cost of advancing their drugs through the trial [19•]. It will be more attractive to pharmaceutical companies if I-SPY 2 can seamlessly incorporate validation of promising agents/combinations for regulatory approval by phase 3 trial. As for the agents/combinations that graduated from the trial, a subsequent phase 3 trial with an appropriate patient population will then be initiated to obtain confirmatory evidence required for FDA approval.
Conclusion
I-SPY 2 is a new generation of clinical trial using a patient-centered, individualized approach, with the goals to tailor the treatment fitting the patient best; it has proven to rapidly select promising agents/combinations for confirmatory phase 3 clinical trial and shorten the duration of drug development and market approval. It already has served as the prototype for new adaptive platform trials for melanoma, glioblastoma, and other diseases such as Alzheimer’s disease. It ultimately benefits breast cancer patients by bringing more innovative and precise options to patients at an earlier stage of breast cancer, where it matters most.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Oliver RT. Regarding: Valero V, Buzdar AU, Hortobagyi GN. Locally advanced breast cancer. The Oncologist 1996;1:8-17. Oncologist. 1996;1(4):278–9.
Tryfonidis K, Senkus E, Cardoso MJ, Cardoso F. Management of locally advanced breast cancer—perspectives and future directions. Nat Rev Clin Oncol. 2015;12(3):147–62. https://doi.org/10.1038/nrclinonc.2015.13.
Newman LA, Mamounas EP. Review of breast cancer clinical trials conducted by the National Surgical Adjuvant Breast Project. Surg Clin North Am. 2007;87(2):279–305, vii. https://doi.org/10.1016/j.suc.2007.02.005.
DiMasi JA, Grabowski HG, Hansen RW. Innovation in the pharmaceutical industry: new estimates of R&D costs. J Health Econ. 2016;47:20–33. https://doi.org/10.1016/j.jhealeco.2016.01.012.
Fisher B, Jeong JH, Bryant J, Anderson S, Dignam J, Fisher ER, et al. Treatment of lymph-node-negative, oestrogen-receptor-positive breast cancer: long-term findings from National Surgical Adjuvant Breast and Bowel Project randomised clinical trials. Lancet. 2004;364(9437):858–68. https://doi.org/10.1016/S0140-6736(04)16981-X.
Sparano JA, Gray RJ, Makower DF, Pritchard KI, Albain KS, Hayes DF, et al. Adjuvant chemotherapy guided by a 21-gene expression assay in breast cancer. N Engl J Med. 2018;379(2):111–21. https://doi.org/10.1056/NEJMoa1804710.
Cardoso F, van’t Veer LJ, Bogaerts J, Slaets L, Viale G, Delaloge S, et al. 70-Gene signature as an aid to treatment decisions in early-stage breast cancer. N Engl J Med. 2016;375(8):717–29. https://doi.org/10.1056/NEJMoa1602253.
DiMasi JA, Hansen RW, Grabowski HG. The price of innovation: new estimates of drug development costs. J Health Econ. 2003;22(2):151–85. https://doi.org/10.1016/s0167-6296(02)00126-1.
Fehrenbacher L, Cecchini R, Geyer C, Rastogi P, Costantino J, Atkins J et al., editors. NSABP B-47 (NRG oncology): phase III randomized trial comparing adjuvant chemotherapy with adriamycin (A) and cyclophosphamide (C) → weekly paclitaxel (WP), or docetaxel (T) and C with or without a year of trastuzumab (H) in women with node-positive or high-risk node-negative invasive breast cancer (IBC) expressing HER2 staining intensity of IHC 1+ or 2+ with negative FISH (HER2-low IBC). San Antonio Breast Cancer Symposium; 2017; San Antonio, Texas: SABCS.
Perez EA, Romond EH, Suman VJ, Jeong JH, Sledge G, Geyer CE Jr, et al. Trastuzumab plus adjuvant chemotherapy for human epidermal growth factor receptor 2-positive breast cancer: planned joint analysis of overall survival from NSABP B-31 and NCCTG N9831. J Clin Oncol. 2014;32(33):3744–52. https://doi.org/10.1200/JCO.2014.55.5730.
•• Park JW, Liu MC, Yee D, Yau C, van’t Veer LJ, Symmans WF, et al. Adaptive randomization of neratinib in early breast cancer. N Engl J Med. 2016;375(1):11–22. https://doi.org/10.1056/NEJMoa1513750First report from I-SPY 2 showing neratinib plus paclitaxel was superior to trastuzumab plus paclitaxel.
Amiri-Kordestani L, Wedam S, Zhang L, Tang S, Tilley A, Ibrahim A, et al. First FDA approval of neoadjuvant therapy for breast cancer: pertuzumab for the treatment of patients with HER2-positive breast cancer. Clin Cancer Res. 2014;20(21):5359–64. https://doi.org/10.1158/1078-0432.CCR-14-1268.
von Minckwitz G, Procter M, de Azambuja E, Zardavas D, Benyunes M, Viale G, et al. Adjuvant pertuzumab and trastuzumab in early HER2-positive breast cancer. N Engl J Med. 2017;377(2):122–31. https://doi.org/10.1056/NEJMoa1703643.
Barker AD, Sigman CC, Kelloff GJ, Hylton NM, Berry DA, Esserman LJ. I-SPY 2: an adaptive breast cancer trial design in the setting of neoadjuvant chemotherapy. Clin Pharmacol Ther. 2009;86(1):97–100.
Cardoso F, Van’t Veer L, Rutgers E, Loi S, Mook S, Piccart-Gebhart MJ. Clinical application of the 70-gene profile: the MINDACT trial. J Clin Oncol. 2008;26(5):729–35. https://doi.org/10.1200/JCO.2007.14.3222.
Berry DA. Bayesian clinical trials. Nat Rev Drug Discov. 2006;5(1):27–36. https://doi.org/10.1038/nrd1927.
DeMichele A, Berry DA, Zujewski J, Hunsberger S, Rubinstein L, Tomaszewski JE, et al. Developing safety criteria for introducing new agents into neoadjuvant trials. Clin Cancer Res. 2013;19(11):2817–23. https://doi.org/10.1158/1078-0432.CCR-12-2620.
Paoloni M, Lyandres J, Buxton M, Berry D, Esserman L, DeMichele A et al., editors. Abstract P2-11-02: a longitudinal look at toxicity management within a platform trial: lessons from the I-SPY 2 TRIAL 2017 February 15 2017; Cancer Res
• Das S, Lo AW. Re-inventing drug development: a case study of the I-SPY 2 breast cancer clinical trials program. Contemp Clin Trials. 2017;62:168–/. https://doi.org/10.1016/j.cct.2017.09.002Outside review of the innovations contained within I-SPY 2.
Rastogi P, Anderson SJ, Bear HD, Geyer CE, Kahlenberg MS, Robidoux A, et al. Preoperative chemotherapy: updates of National Surgical Adjuvant Breast and Bowel Project protocols B-18 and B-27. J Clin Oncol. 2008;26(5):778–85.
Wolmark N, Wang J, Mamounas E, Bryant J, Fisher B. Preoperative chemotherapy in patients with operable breast cancer: nine-year results from National Surgical Adjuvant Breast and Bowel Project B-18. J Natl Cancer Inst Monogr. 2001;30:96–102.
Cortazar P, Zhang L, Untch M, Mehta K, Costantino JP, Wolmark N, et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet. 2014;384(9938):164–72. https://doi.org/10.1016/S0140-6736(13)62422-8.
Administration FDA. Guidance for industry: pathological complete response in neoadjuvant treatment of high-risk early-stage breast cancer: use as an endpoint to support accelerated approval.: fda.gov; October 2014.
Berry DA, Hudis CA. Neoadjuvant therapy in breast cancer as a basis for drug approval. JAMA Oncol. 2015;1(7):875–6. https://doi.org/10.1001/jamaoncol.2015.1293.
Esserman LJ, Berry DA, DeMichele A, Carey L, Davis SE, Buxton M, et al. Pathologic complete response predicts recurrence-free survival more effectively by cancer subset: results from the I-SPY 1 TRIAL—CALGB 150007/150012, ACRIN 6657. J Clin Oncol. 2012;30(26):3242–9. https://doi.org/10.1200/JCO.2011.39.2779.
• Yee D, DeMichele A, Isaacs C, Symmans F, Yau C, Albain KS et al., editors. Pathological complete response predicts event-free and distant disease-free survival in the I-SPY2 TRIAL. San Antonio Breast Cancer Symposium; 2017; San Antonio, Texas: SABCS. I-SPY 2 report of the association between pCR and event-free and distant disease-free survival.
Freidlin B, Korn EL, Gray R, Martin A. Multi-arm clinical trials of new agents: some design considerations. Clinical Cancer Research: an official journal of the American Association for Cancer Research. 2008;14(14):4368-4371. doi:https://doi.org/10.1158/1078-0432.CCR-08-0325.
Korn EL, Freidlin B. Adaptive clinical trials: advantages and disadvantages of various adaptive design elements. J Natl Cancer Inst. 2017;109(6). https://doi.org/10.1093/jnci/djx013.
• Wolf DM, Yau C, Sanil A, Glas A, Petricoin E, Wulfkuhle J, et al. DNA repair deficiency biomarkers and the 70-gene ultra-high risk signature as predictors of veliparib/carboplatin response in the I-SPY 2 breast cancer trial. NPJ Breast Cancer. 2017;3:31. https://doi.org/10.1038/s41523-017-0025-7Biomarker analysis from I-SPY 2 identifies potential signatures associated with improved response to carboplatin-containing regimens.
Castano Z, Marsh T, Tadipatri R, Kuznetsov HS, Al-Shahrour F, Paktinat M, et al. Stromal EGF and IGF-I together modulate plasticity of disseminated triple-negative breast tumors. Cancer Discov. 2013;3(8):922–35. https://doi.org/10.1158/2159-8290.CD-13-0041.
Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, Huang S, et al. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008;68(3):826–33.
Deeks ED. Neratinib: first global approval. Drugs. 2017;77(15):1695–704. https://doi.org/10.1007/s40265-017-0811-4.
• Rugo HS, Olopade OI, DeMichele A, Yau C, van’t Veer LJ, Buxton MB, et al. Adaptive randomization of veliparib-carboplatin treatment in breast cancer. N Engl J Med. 2016;375(1):23–34. https://doi.org/10.1056/NEJMoa1513749First I-SPY 2 report of veliparib, carboplatin, and paclitaxel improving pCR rates.
• Loibl S, O’Shaughnessy J, Untch M, Sikov WM, Rugo HS, McKee MD, et al. Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): a randomised, phase 3 trial. Lancet Oncol. 2018;19(4):497–509. https://doi.org/10.1016/S1470-2045(18)30111-6Phase 3 randomized trial validating findings from I-SPY 2 (ref. 33). Trial showed improved pCR rates were due to carboplatin but not veliparib.
Severson TM, Wolf DM, Yau C, Peeters J, Wehkam D, Schouten PC, et al. The BRCA1ness signature is associated significantly with response to PARP inhibitor treatment versus control in the I-SPY 2 randomized neoadjuvant setting. Breast Cancer Res. 2017;19(1):99–9. https://doi.org/10.1186/s13058-017-0861-2.
Swain SM, Baselga J, Kim SB, Ro J, Semiglazov V, Campone M, et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med. 2015;372(8):724–34. https://doi.org/10.1056/NEJMoa1413513.
Buxton M, DeMichele AM, Chia S, van’t Veer L, Chien J, Wallace A et al., editors. Abstract CT106: efficacy of pertuzumab/trastuzumab/paclitaxel over standard trastuzumab/paclitaxel therapy for HER2+ breast cancer: results from the neoadjuvant I-SPY 2 TRIAL 2016: AACR.
DeMichele AM, Moulder S, Buxton M, Yee D, Wallace A, Chien J et al., editors. Abstract CT042: efficacy of T-DM1+ pertuzumab over standard therapy for HER2+ breast cancer: results from the neoadjuvant I-SPY 2 TRIAL 2016: AACR.
Schott AF, Hayes DF. Defining the benefits of neoadjuvant chemotherapy for breast cancer. J Clin Oncol. vol 15. United States 2012. p. 1747-1749.
Alexander BM, Wen PY, Trippa L, Reardon DA, Yung WK, Parmigiani G, et al. Biomarker-based adaptive trials for patients with glioblastoma—lessons from I-SPY 2. Neuro-Oncology. 2013;15(8):972–8. https://doi.org/10.1093/neuonc/not088.
Messmer MF, Wilhelm EE, Shoulson I. I-SPY 2 breast cancer trial as a model for innovation in Alzheimer disease therapies. JAMA Neurol. 2017;74(9):1027–8. https://doi.org/10.1001/jamaneurol.2017.1528.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Douglas Yee reports grants from QuantumLeap, grants from the National Cancer Institute (P30-CA077598), and grants from the National Cancer Institute (P01 CA210961) during the conduct of the study. Dr. Yee also reports personal fees from AstraZeneca and Puma outside the submitted work. Haiyun Wang declares no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Clinical Trials
Rights and permissions
About this article

Cite this article
Wang, H., Yee, D. I-SPY 2: a Neoadjuvant Adaptive Clinical Trial Designed to Improve Outcomes in High-Risk Breast Cancer. Curr Breast Cancer Rep 11, 303–310 (2019). https://doi.org/10.1007/s12609-019-00334-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12609-019-00334-2