
Abstract
Silicon is one of the most promising anode materials for lithium-ion batteries (LIBs), but it suffers from pulverization and hence poor cycling stability due to the large volume variation during lithiation/delithiation. The core–shell structure is considered as an effective strategy to solve the expansion problem of silicon-based anodes. In this paper, the double-shell structured Si@SnO2@C nanocomposite with nano-silicon as the core and SnO2, C as the shells is synthesized by a facile hydrothermal method. Structural characterization shows that Si@SnO2@C nanocomposite is composed of crystalline Si, crystalline SnO2 and amorphous C, and the contents of them are 42.1 wt%, 37.8 wt% and 20.1 wt%, respectively. Transmission electron microscope (TEM) observations confirm the double-shell structure of Si@SnO2@C nanocomposite, and the thicknesses of the SnO2 and C layers are 20 and 7 nm. The Si@SnO2@C electrode exhibits a high initial discharge capacity of 2777 mAh·g−1 at 100 mA·g−1 and an excellent rate capability of 340 mAh·g−1 at 1500 mA·g−1. The outstanding capacity retention is 50.2% after 300 cycles over a potential of 0.01 to 2.00 V (vs. Li/Li+) at 500 mA·g−1. The resistance of solid electrolyte interphase (SEI) film (Rf) and charge transfer resistance (Rct) of Si@SnO2@C are 7.68 and 0.82 Ω, which are relatively smaller than those of Si@C (21.64 and 2.62 Ω). It is obviously seen that the SnO2 shell can reduce the charge transfer resistance, leading to high ion and electron transport efficiency in the Si@SnO2@C electrode. The incorporation of SnO2 shell is attributed to the enhanced rate capability and cycling performance of Si@SnO2@C nanocomposite.
Graphical abstract

摘要
硅是锂离子电池极具应用前景的负极材料之一, 但其在脱嵌锂过程中存在体积变化大的问题, 导致硅负极材料发生粉化, 使得电池的循环稳定性下降。核壳结构被认为是解决硅基负极体积膨胀问题的有效策略。本文采用简单的水热法制备了以纳米硅为核心、二氧化锡和碳为壳层的具有双层壳结构Si@SnO2@C纳米复合材料。结构表征表明, Si@SnO2@C纳米复合材料由晶体Si、晶体SnO2和非晶C组成, 其含量分别为42.1 wt%、37.8 wt%和20.1 wt%。透射电镜结果证实了Si@SnO2@C纳米复合材料具有双壳层结构, SnO2和C壳层的厚度分别为20和7 nm。经过电化学循环测试, Si@SnO2@C电极在100 mA g−1电流密度下具有2777 mAh g−1的初始放电容量, 在电流密度为1500mA g−1时表现出340 mAh g−1的优异倍率能力。在电流密度为500 mA g−1下, 在0.01到2.00 V(对Li/Li+)的电压范围内进行300次循环后, 容量保持率高达50.2%。通过EIS测试, 计算得Si@SnO2@C的SEI膜电阻(Rf)和电荷转移电阻(Rct)分别为7.68和0.82 Ω, 其值远小于 Si@C (21.64和2.62 Ω)。对比发现, SnO2壳层可以降低电荷转移电阻, 使得 Si@SnO2@C电极表现出较高的离子和电子传输效率, 大大提高了Si@SnO2@C纳米复合材料的倍率性能和循环性能。
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Lithium-ion batteries (LIBs) are promising high-efficiency secondary batteries used for portable electronic and electric vehicles [1, 2]. Commercial LIBs employing graphite anodes cannot meet the growing energy demand due to their low theoretical capacity (372 mAh·g–1) and energy density (1–10 Wh·kg–1) [3]. In recent years, great efforts have been devoted to exploiting new anode materials with high-capacity, long cyclic performance and low cost, such as silicon [4], tin [5] and metal oxide [6,7,8], and so on. Among these, silicon is considered as an ideal substitute for graphite materials due to its high-capacity (4200 mAh·g−1), low potential (370 mV (vs. Li/Li+)) [9], and natural abundance [10]. Although it has high specific capacity, the practical application of silicon-based material in LIBs is restricted because of the large volume change (300%–400%) during the charge–discharge process. It may lead to the pulverization of silicon [11], contact failure between the active material and the current collector as well as the unstable solid electrolyte interphase (SEI) film [12]. Furthermore, the poor intrinsic conductivity of silicon combined with the reduced electrochemical activity results in unsatisfactory charging rate capability [13, 14].
According to studies, constructing nanostructure and compositing silicon with other materials could effectively improve the electrochemical performance of silicon by buffering the volume expansion during the charge–discharge processes [15] and could promote the conductivity of silicon [16]. There are many successful cases of synthesizing the silicon-based nanostructured composites, such as yolk/double-shell silicon-based composite [17, 18], ant-nest-like bulk porous silicon [19], hollow porous silicon microspheres [20] and carbon nanotube@silicon core–shell wires [21]. Among the various materials, Sn is an excellent active material owing to its superior electrical conductivity, moderate potential and abundant reserves [22, 23]. It can not only buffer the volume deformation of Si, but also improve the dispersion of Si because of its high ductility [24]. For instance, Lee et al. [25] designed a new ternary composite anode material using core-cut nozzle technology. Si and Sn nanoparticles were separated in double-hole carbon nanofibers (Si-Sn-DHCNFs). The specific capacity of Si-Sn-DHCNFs was 1000 mAh·g–1 at 100 mA·g–1, and rate performance was 720 mAh·g–1 at 10,000 mA·g–1. Yang et al. [26] fabricated a silicon/tin@amorphous carbon-graphite (Si/Sn@C-G) composite by high-energy ball milling. The initial Coulombic efficiency of Si/Sn@C-G was as high as 81.5%. Even after 100 cycles, Si/Sn@C-G composite still exhibited a stable reversible capacity of 612.6 mAh·g–1. The results of cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) demonstrated that Sn could be used as an active matrix to significantly reduce the polarization resistance [27].
Although the introduction of Sn can improve the conductivity and electrochemical performance of Si [24, 28], the large volume change of Sn may cause pulverization, side reactions, as well as SEI propagation, further deteriorating the contact [5]. In addition, owing to the low melting temperature of Sn, it is easy to form clusters and two-phase regions. Especially in the case of compounding of Sn and C, the high temperature heat-treatment required during carbonization results in the agglomeration of molten Sn, leading to volume mismatch during the cycling process [29]. The core–shell structure is constructed using SnO2 with high melting point and hardness to replace Sn compounding with Si, and it may improve the conductivity and capacity while reducing the generation of Sn clusters [26]. More importantly, Li2O (SnO2 + 4Li+ + 4e− ↔ Sn + 2Li2O) formed by SnO2 in the conversion reaction is expected to help alleviating the expansion stress during alloying (Sn + 4.4Li+ + 4.4e− ↔ Li4.4Sn) [5]. In recent years, reports have highlighted the beneficial effects of the combination of Si and SnO2 on the storage performance of lithium [30]. For example, Zhou et al. [31] proposed a facile self-assembly method to decorate silicon hollow nanospheres with SnO2 nanowires. The results showed that the Si@SnO2 core–shell structure exhibited a remarkable synergistic effect in high-capacity reversible lithium storage, delivering a high reversible capacity of 1869 mAh·g–1 at 500 mA·g–1 after 100 cycles. Ma et al. [32] designed SnO2@Si nanospheres with a novel hollow structure. It not only had high volumetric capacity as anode of LIBs, but also protected Sn aggregation and suppressed SEI thickening. The above results demonstrated that Si-SnO2 based composites had excellent electrochemical kinetics and lithium storage properties as potential anode materials. Despite the great achievements, extensive SEI formed on the high interface area of SnO2 in the first discharge process when SnO2 contacted with the electrolyte directly, and this resulted in the high irreversible capacity loss [33]. Therefore, further work is required to design and optimize the microstructures of high-capacity electrodes to make them stable during the lithiation-delithiation process.
Although Si-Sn-C composite anode has shown improvement in the electrochemical performance, Sn is mostly composited with Si-C materials for its low melting point. As a result, Si-Sn-C composites still have problems such as volume expansion, poor cycle stability, and low Coulombic efficiency, which are the main reasons affecting their electrochemical performance. In this work, we constructed a double-shelled structured Si@SnO2@C nanocomposite. As an intermediate layer between Si and C, the SnO2 shell may improve electrochemical reaction dynamics. Because intermediate layer allows the nanocomposite to form a conductive network from inside to outside, it can effectively facilitate the transportation of lithium ions and electrons between the active material and the electrolyte. SnO2 is reduced to a ductile Sn during the electrochemical reaction, which can effectively relieve the surface tension during the lithium process. The outermost carbon shell can prevent the contact between silicon, SnO2 and electrolyte, improving the structural stability of the material. This structure is expected to provide the new ideas for the improvement of silicon-based composite for LIBs anode.
2 Experimental
2.1 Preparation of Si@SnO2@C nanocomposite
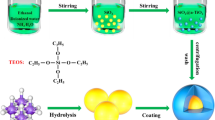
Figure 1 shows a schematic diagram of the preparation of Si@SnO2@C nanocomposite. Si@SnO2@C was prepared by a two-step hydrothermal process. The core–shell structured Si@SnO2 nanocomposite was prepared in the first step. In a typical experiment, a clear solution was obtained by dissolving 0.848 g Na2SnO3 and 7.2 g urea in 136 ml of deionized water. 72 ml ethanol was then added to the solution. Next, 0.52 g silicon nanopowder was added to the solution and dispersed by ultrasonication for 10 min. The resulting dispersion was placed in a 300 ml teflon-lined autoclave and heated in an airflow electric oven at a temperature of 165 °C for 12 h. Si@SnO2 powder was collected by centrifugation, washed with deionized ethanol and water, and dried under vacuum at 80 °C overnight. The double-shell structured Si@SnO2@C nanocomposite was prepared afterward. Typically, 1.5 g glucose was dissolved in 50 ml deionized water to make a precursor solution. Then, 1.0 g Si@SnO2 nanocomposite was added to the precursor solution and ultrasonically dispersed for 10 min before the mixture was hydrothermally treated in a teflon-lined autoclave at 190 °C for 12 h to form the Si@SnO2@C nanocomposite. Finally, a carbonization treatment was carried out at 500 °C for 3 h in N2 atmosphere. For comparison, Si@C nanocomposite was prepared under the same experimental conditions.

Schematic illustration of preparation of Si@SnO2@C nanocomposite
2.2 Characterization
The microstructural characterization of the nanocomposites was conducted by scanning electron microscopy (SEM, Hitachi S-4800) and transmission electron microscopy (TEM, Tecnai G2 F30). Thermogravimetric analysis (TG, TGA Q500) was performed with flowing air at temperatures up to 900 °C (10 °C·min–1 heating ramp) to determine the carbon content in nanocomposite. Inductively coupled plasma-optical emission spectrometer (ICP-OES) was used to measure the elemental contents of samples (Spectro Genesis ICP). The phases of nanocomposite were identified by an X-ray diffractometer (Rigaku D/MAX) at 2500 V.
2.3 Electrochemical measurements
The coin cells (CR 2032) were used to test the electrochemical performance of Si@SnO2@C nanocomposite. The electrodes were prepared by mixing Si@SnO2@C nanocomposite, binder (carboxymethylcellulose sodium/ polymerized styrene butadiene rubber, CMC/SBR) and conductive carbon (super P) with weight ratio of 8:1:1. The electrolyte consists of 1 mol·L–1 LiPF6 in ethylene carbonate (EC), dimethyl carbonate (DMC), and ethyl methyl carbonate (EMC) with volume ratio of 2:6:2. Lithium metal was as the counter electrode of the coin-type (CR 2032) cells with a glass fiber membrane as the separator. The process of assembling is in an argon-filled glove box. The Land battery system (CT2001A) was used to test the galvanostatic charge–discharge (GCD) measurements at a constant current density in a voltage window of 0.01–2.00 V (vs. Li/Li+). CV and EIS test were carried out by CHI600E electrochemical workstation.
3 Results and discussion
Figure 2a shows XRD patterns of Si, Si@C, Si@SnO2 and Si@SnO2@C powders. For all the four samples, the diffraction peaks at 2θ of 28.4°, 47.3°, 56.1°, 56.6°, 68.9° and 76.3° are indexed to the (111), (220), (311), (400), (331) and (422) planes of Si (JCPDS No. 27-1402) [34, 35]. As for Si@SnO2 and Si@SnO2@C nanocomposites, the weak diffraction peaks at 2θ of 26.6°, 33.9° and 51.8° are indexed to the (110), (101) and (211) planes of SnO2 (JCPDS No. 41-1445) [36]. The synthesized SnO2 has an average crystalline size of 5.23 nm, as calculated by the Debye–Scherrer formula. No peaks from carbon are observed for Si@C and Si@SnO2@C, and it indicates that the C shell is amorphous [37].

a XRD patterns of Si, Si@C, Si@SnO2 and Si@SnO2@C; b TG curves of Si@C, Si@SnO2 and Si@SnO2@C
TG analysis is performed to determine the contents of C for Si@C and Si@SnO2@C nanocomposites, as shown in Fig. 2b. The weight loss below 100 °C is 2.5 wt%, which is attributed to the removal of physical adsorbed water in powders [33]. The weight loss in the temperature range of 100–600 °C is attributed to carbon oxidation [20]. Based on the TG results, the C contents of Si@C and Si@SnO2@C are about 24.0 wt% and 20.1 wt%, respectively. The weight gain beyond 600 °C maybe due to the oxidation of silicon [38]. In addition, ICP-OES test of the remaining solution after hydrothermal synthesis of Si@SnO2 showed that the contents of elemental Si and Sn were less than 3 × 10–6. ICP results indicate that Na2SnO3 is converted into SnO2. Therefore, the contents of Si, SnO2 and C in the Si@SnO2@C nanocomposite are calculated as about 40.7 wt%, 39.2 wt% and 20.1 wt%, respectively.
Figure 3 shows SEM images of Si, Si@C, Si@SnO2 and Si@SnO2@C. The particle sizes of Si powders are in the range of 50–100 nm. There is no significant microstructural difference between Si and Si@C powders, indicating the relatively low layer thickness of carbon in Si@C nanocomposite. The surface of Si@SnO2 seems to be rougher than that of the Si particles. This phenomenon suggests that there is a layer made of SnO2 nanoparticles being formed on the surface of Si particles. The surface of Si@SnO2@C nanocomposite is the roughest among these powders, and amorphous carbon forms a carbon layer on the rough surface of Si@SnO2 particles. According to the theory of hydrothermal growth [39, 40], SnO2 and C nanoparticles were assembled on the particle surface through the Ostwald ripening process [41]. During the hydrothermal process, Na2SnO3 decomposes to form SnO2 [42]. In order to decrease the total energy of the system, the generated SnO2 nanoparticles spontaneously form a uniform shell on the surface of the silicon particles, which results in the formation of Si@SnO2. Similarly, C is coated on the surface of Si@SnO2 particles to form a Si@SnO2@C double-shell structure.

SEM images of a Si, b Si@C, c Si@SnO2, d Si@SnO2@C
The microstructure of Si@SnO2@C nanocomposite is characterized by TEM, as shown in Fig. 4. The double-shell structure of Si@SnO2@C can be clearly discerned under the scanning TEM mode in Fig. 4a. In Fig. 4b, the thicknesses of carbon and SnO2 shell is determined to be ~ 20 and ~ 7 nm. The carbon shell is found to be amorphous, which is identified with XRD results. In addition, HRTEM images of Si@SnO2@C show the distinct crystal lattice fringes with interplanar spacings of 3.35 and 3.13 nm, which belong to the (110) plane of SnO2 and (111) plane of Si [31]. Figure 4c–g shows EDS mapping results of the spatial distributions of Si, O, Sn and C elements, which further proves the double-shell structure of Si@SnO2@C nanocomposite.

Images of Si@SnO2@C: a TEM image, b HRTEM image, c–g EDS element mappings of marked area
The electrochemical performance of the synthesized nanocomposites as LIBs anode is evaluated by GCD measurements using half cells. The rate capacity of the electrode is tested in the voltage range of 0.01–2.00 V (vs. Li+/Li). Figure 5a illustrates the rate capacity of the Si@C and Si@SnO2@C electrodes at different current densities from 100 to 1500 mA·g–1 and then back to 100 mA·g–1. The rate performance of the Si@SnO2@C electrode shows a significant improvement compared to Si@C. At current densities of 100, 200, 500, 1000 and 1500 mA·g–1, the reversible capacities of Si@SnO2@C nanocomposite are 1098, 875, 701, 515 and 340 mAh·g–1. After 50 cycles, the capacity remains 919 mAh·g–1 at 100 mA·g–1, and the capacity recovery rate reaches 84%, demonstrating its outstanding reversibility and cycle stability [31]. In the meantime, the capacities of Si@C anode are only 249, 200, 158, 125 and 106 mAh·g–1 at current densities of 100, 200, 500, 1000 and 1500 mA·g–1. To further analyze its rate performance, Fig. 5b shows the charge–discharge curves of the Si@SnO2@C electrode at different current densities. Even at a high current density of 1500 mA·g–1, the charging and discharging plateaus have no obvious hysteresis growth, demonstrating the superior electronic/ionic transport properties of the Si@SnO2@C anode [43].

a Rate performance of Si@C and Si@SnO2@C electrodes at various current densities; b GCD profiles of Si@SnO2@C electrode at increasing discharge current density from 100 to 1500 mA·g−1; c cycling performance and Coulombic efficiency of Si@SnO2@C and Si@C anodes at 500 mA·g–1 for 300 cycles (100 mA·g–1 for the first five cycles)
Figure 5c shows the stabilities of the Si@SnO2@C and Si@C anodes during the charge–discharge process. The galvanostatic cycling performances of Si@SnO2@C and Si@C anodes were tested at 500 mA·g–1 in the voltage range of 0.01–2.00 V (vs. Li+/Li) for 300 cycles (100 mA·g–1 for the first five cycles). In the initial discharge, the Si@SnO2@C electrode exhibits a high discharge capacity of 2777.5 mAh·g–1. The capacity is retained 554.3 mAh·g–1 after 300 charge–discharge cycles, showing outstanding capacity retention of 50.2% of its highest discharge capacity of 1104.2 mAh·g–1 at 500 mA·g–1. Furthermore, the Coulombic efficiency of the Si@SnO2@C electrode exhibits a significant increase in initial cycles and maintains approximately 98% after six cycles. The cycling figure of the Si@C electrode is similar to that of the carbon anodes. The reason of the poor electrochemical performance of Si@C electrodes still needs to be further explored.
The electrochemical performance of the nanocomposite and the changes of surface morphology during cycling processes are further investigated by SEM characterization. Figure 6 shows SEM images of the Si@C and Si@SnO2@C electrodes before and after 300 charge–discharge cycles at 500 mA·g–1. Si@SnO2@C and Si@C particles maintain the spherical shape after cycling, and there are almost no agglomerations and SEI blocks on the surface. This is mainly due to the protection of the carbon outermost layer. The results show that the Si@SnO2@C anode has good stability and the double-shell structure can help withstand the mechanical stress during the repeated electrochemical cycles. Both Si@SnO2@C and Si@C materials undergo volume expansion which causes the increase of the impedance of the electrode, since the conductivity between the active materials and the copper is gradually decreasing as cycling proceeds [43,44,45,46]. The structural integrity of electrode is maintained after long-term electrochemical cycling tests. Thus, the core–shell structure of nanocomposites is beneficial for the electrode–electrolyte interface stability for long-term cycling and stable electrical energy delivery. The stable reversible reaction between Li+ and the active material in the electrode in each cycle is guaranteed.

SEM images of Si@C electrodes a before and c after 300 charge–discharge cycles at 500 mA·g−1; SEM images of Si@SnO2@C electrodes b before and d after 300 charge–discharge cycles at 500 mA·g−1
Figure 7 shows typical SEM images of the cross sections of the Si@C and Si@SnO2@C anodes before and after 300 charge–discharge cycles. All electrodes maintain good integrity despite thickening of both electrode sheets after 300 cycles. The thickness of Si@C electrode increases from 16.2 to 19.4 μm. Combined with the electrochemical performance of the Si@C electrode, it further confirms that the silicon in the Si@C electrode does not participate in the electrochemical reaction. Compared with the reported Si-Sn based electrodes [17, 26, 47], the double-shell structure of Si@SnO2@C can effectively alleviate the volume expansion, and the thickness of electrode is increased by 90.57% after 300 cycles, which is mainly attributed to the dual protection of the high reactive SnO2 interlayer and uniform carbon outer layer.

Cross-sectional SEM images of electrode layers of Si@C: a before and c after 300 cycles; Si@SnO2@C: b before and d after 300 cycles
Figure 8 shows CV curves of Si@SnO2@C and Si@C electrodes at a scan rate of 1 mV·s−1. As for Si@SnO2@C electrode, in the first cathodic (lithium insertion) scan, the reduction peak at 0.9 V is attributed to the formation of Li2O and SEI. It disappears from the subsequent cycles as a result of stability of the SEI film [43]. The electrode exhibits one discernible reduction peak near 0 V, corresponding to the superposition reactions of Si, Sn with Li. To be specific, the process involves Li+ intercalation with Si and Sn to form amorphous LixSi alloy [34] and LixSn alloy [20]. The reduction peak at 1.3 V is due to Li+ intercalation into Sn [5]. During delithiation processes, the oxidation peak at 0.35 V is related to the delithiation reaction of Li-Si alloy, and the peak at 0.7 V is caused by the delithiation process of LixSn and LixSi alloy [46, 48]. The two weak peaks at 1.26 and 1.87 V correspond to the oxidation reaction of Sn to SnO and SnO to SnO2, respectively [43]. With continuous cycling, the shapes of CV profiles remain similar, except for a slight shift and gradual broadening of the redox peak. It shows that the electrode active material is gradually activated, and more active electrode materials participate with the progress of cycling [5]. The reason for the large capacity loss in the first three cycles is the irreversible electrochemical reactions during the initial discharge, the reaction between the super P and electrolyte decomposition [31]. After the first five cycles, all the additional CV curves almost coincide, indicating that the lithiation and delithiation reactions for the Si@SnO2@C nanocomposite are highly reversible [33].

CV curves at a scan rate of 1 mV·s−1 in range of 0.01–3.00 V (vs. Li/Li+): Si@SnO2@C nanocomposite at a the first 5 cycles and b 6–10 cycles; Si@C nanocomposite at c the first 5 cycles and d 6–10 cycles
The electrochemical reactions of Si@SnO2@C nanocomposite are as follows [42, 49]:
On the basis of the above results, it can be concluded that the introduction of SnO2 into the silicon-based composite effectively enhances both the electronic conductivity and the ionic diffusion efficiency in the anode [42]. However, SnO2 forms the irreversible phase (Li2O) during the first lithiation, causing the formation of dead lithium and a poor Coulombic efficiency [47, 50]. This is probably the main reason that the Coulombic efficiency is low for Si@SnO2@C nanocomposite at early cyclings. As a comparison, CV measurement of Si@C is also implemented as shown in Fig. 8. The coincidence of the curves is much poorer compared with that of Si@SnO2@C, indicating a severe polarization in Si@C and resulting in a poor reversible stability [51]. These consequences provide evidence for a more stable reversibility of Si@SnO2@C.
To investigate the impedance changes of the Si@SnO2@C nanocomposite, EIS measurement is used to perform with the new cells and the cells after 3 galvanostatic charge–discharge cycles at the current density of 500 mA·g–1 for Si@C and Si@SnO2@C nanocomposites. The Nyquist plots of Si@C and Si@SnO2@C cells are shown in Fig. 9. All curves consist of two suppressed semicircles at high frequencies and a slope at low frequencies corresponding to the SEI resistance (Rf) [52], the charge transfer resistance (Rct) and the slash at low frequencies is called Warburg impedance [53]. The inset in Fig. 9 is the equivalent circuit in the form of Rs (CPE1, Rf) (CPE2, Rct) Wo [54]. Rs is the resistance of the cell. CPE1 and Rf are the charge transfer capacitance and resistance. CPE2 and Rct are the capacitance and resistance of Li+ transition through the SEI. Wo represents the Warburg impedance associated with the diffusion of Li+ into the bulk electrodes [55]. The data are fitted to the equivalent circuit and the fitted values are reported in Table 1. The Rf and Rct values of Si@SnO2@C before cycling are 7.68 and 0.82 Ω, which are smaller than those of Si@C (21.64 and 2.62 Ω). The result implies that the architecture of Si@SnO2@C facilitates the transfer of charge on the interface [56]. The introduction of SnO2 can greatly improve the conductivity of the silicon anode and enhance the connection between the active material and the current collector. The Rct and Rf values of Si@SnO2@C after 3 cycles are close to those before cycling, indicating that the double-shell structure is conductive to the formation of a stable SEI and facilitates electrochemical reaction dynamics. On the contrary, the Rct value of Si@C electrode increases significantly after cycling. Thus, the EIS results show that Si@SnO2@C electrode exhibits an excellent stability during cycling.

Nyquist plots of a Si@C and b Si@SnO2@C nanocomposites after initial and the 3rd cycles
4 Conclusion
In summary, the double-shell structured Si@SnO2@C nanocomposite has been prepared successfully by a two-step hydrothermal method. Si@SnO2@C nanocomposite is composed of crystalline Si, crystalline SnO2 and amorphous C, and the thicknesses of SnO2 and C layers are 20 and 7 nm, respectively. The nanostructured Si@SnO2@C anode exhibits excellent specific capacity, rate capability and high electrical conductivity. The initial capacity of Si@SnO2@C nanocomposite is as high as 2777.48 mAh·g–1 and retains 544.3 mAh·g−1 after 300 cycles. Si@SnO2@C nanocomposite delivers a high capacity of 1098, 875, 701, 515 and 340 mAh·g–1 at 100, 200, 500, 1000 and 1500 mA·g–1. The outstanding electrochemical performance of Si@SnO2@C is owing to the SnO2 shell which generates Sn in the electrochemical reaction and acts as a conductive network during cycling. This Sn can reduce the charge transfer resistance and thus lead to high ion and electron transport efficiency. Considering the high abundance of Si and SnO2, Si@SnO2@C may serve as a high-energy–density anode material in Li-ion batteries.
References
Lu J, Chen Z, Ma Z, Pan F, Curtiss LA, Amine K. The role of nanotechnology in the development of battery materials for electric vehicles. Nat Nanotechnol. 2016;11(12):1031. https://doi.org/10.1038/nnano.2016.207.
Manj RZA, Zhang F, Rehman WU, Luo W, Yang J. Toward understanding the interaction within silicon-based anodes for stable lithium storage. Chem Eng J. 2020;385:123821. https://doi.org/10.1016/j.cej.2019.123821.
Holzapfel M, Buqa H, Scheifele W, Novak P, Petrat FM. A new type of nano-sized silicon/carbon composite electrode for reversible lithium insertion. Chem Commun. 2005;12:1566. https://doi.org/10.1039/b417492e.
Lee SJ, Kim HJ, Hwang TH, Choi S, Park SH, Deniz E, Jung DS, Choi JW. Delicate structural control of Si-SiOx-C composite via high-speed spray pyrolysis for Li-ion battery anodes. Nano Lett. 2017;17(3):1870. https://doi.org/10.1021/acs.nanolett.6b05191.
Chen Y, Ge D, Zhang J, Chu R, Zheng J, Wu C, Zeng Y, Zhang Y, Guo H. Ultrafine Mo-doped SnO2 nanostructure and derivative Mo-doped Sn/C nanofibers for high-performance lithium-ion batteries. Nanoscale. 2018;10(36):17378. https://doi.org/10.1039/c8nr01195h.
Bhattacharya P, Kota M, Suh DH, Roh KC, Park HS. Biomimetic spider-web-like composites for enhanced rate capability and cycle life of lithium ion battery anodes. Adv Energy Mater. 2017;7(17):1700331. https://doi.org/10.1002/aenm.201700331.
Zhang Y, Lu Y, Feng S, Liu D, Ma Z, Wang S. On-site evolution of ultrafine ZnO nanoparticles from hollow metal-organic frameworks for advanced lithium ion battery anodes. J Mater Chem A. 2017;5(43):22512. https://doi.org/10.1039/c7ta08284c.
Zhang Y, Chen P, Gao X, Wang B, Liu H, Wu H, Liu H, Dou S. Nitrogen-doped graphene ribbon assembled core-sheath MnO@Graphene scrolls as hierarchically ordered 3D porous electrodes for fast and durable lithium storage. Adv Funct Mater. 2016;26(43):7754. https://doi.org/10.1002/adfm.201603716.
Poizot P, Laruelle S, Grugeon S, Dupont L, Tarascon JM. Searching for new anode materials for the Li-ion technology: time to deviate from the usual path. J Power Sour. 2001;SI(97–98):235. https://doi.org/10.1016/S0378-7753(01)00508-0.
Huggins R. Lithium alloy negative electrodes. J Power Sour. 1999;81(2):13. https://doi.org/10.1016/S0378-7753(99)00124-X.
Shang H, Zuo Z, Yu L, Wang F, He F, Li Y. Low-temperature growth of all-carbon graphdiyne on a silicon anode for high-performance lithium-ion batteries. Adv Mater. 2018;30(27):e1801459. https://doi.org/10.1002/adma.201801459.
Ruffo R, Hong SS, Chan CK, Huggins RA, Cui Y. Impedance analysis of silicon nanowire lithium ion battery anodes. J Phys Chem. 2009;113(26):11390. https://doi.org/10.1021/jp901594g.
Wang Q, Meng T, Li Y, Yang J, Huang B, Ou S, Meng C, Zhang S, Tong Y. Consecutive chemical bonds reconstructing surface structure of silicon anode for high-performance lithium-ion battery. Energy Storage Mater. 2021;39:354. https://doi.org/10.1016/j.ensm.2021.04.043.
Zhang M, Li J, Sun C, Wang Z, Li Y, Zhang D. Durable flexible dual-layer and free-standing silicon/carbon composite anode for lithium-ion batteries. J. Alloys Compd. 2022;932:167687. https://doi.org/10.1016/j.jallcom.2022.167687.
Su L, Jing Y, Zhou Z. Li ion battery materials with core-shell nanostructures. Nanoscale. 2011;3(10):3967. https://doi.org/10.1039/c1nr10550g.
Wu Z, Luo J, Peng J, Liu H, Chang B, Wang X. Rational architecture design of yolk/double-shells Si-based anode material with double buffering carbon layers for high performance lithium-ion battery. Green Energy Environ. 2021;6(4):517. https://doi.org/10.1016/j.gee.2020.06.009.
Xiao X, Wang JS, Liu P, Sachdev AK, Verbrugge MW, Haddad D, Balogh MP. Phase-separated silicon-tin nanocomposites for high capacity negative electrodes in lithium ion batteries. J Power Sour. 2012;214:258. https://doi.org/10.1016/j.jpowsour.2012.04.028.
Lu W, Guo X, Luo Y, Li Q, Zhu R, Pang H. Core-shell materials for advanced batteries. Chem Eng J. 2019;355:208. https://doi.org/10.1016/j.cej.2018.08.132.
An W, Gao B, Mei S, Xiang B, Fu J, Wang L, Zhang Q, Chu PK, Huo K. Scalable synthesis of ant-nest-like bulk porous silicon for high-performance lithium-ion battery anodes. Nat Commun. 2019. https://doi.org/10.1038/s41467-019-09510-5.
Chao Z, Zhao L, Ze Z, Cai J, Yang Z, Ji Y. Synthesis of the SnO2@C@GN hollow porous microspheres with superior cyclability for Li-ion batteries. Chem Phys Lett. 2021. https://doi.org/10.1016/j.cplett.2021.138566.
Fan Y, Zhang Q, Lu C, Xiao Q, Wang X, Tay BK. High performance carbon nanotube-Si core-shell wires with a rationally structured core for lithium ion battery anodes. Nanoscale. 2013;5(4):1503. https://doi.org/10.1039/c3nr33683b.
Beaulieu LY, Hatchard TD, Bonakdarpour A, Fleischauer MD, Dahn JR. Reaction of Li with alloy thin films studied by in situ AFM. J Electrochem Soc. 2003;150(11):A1457. https://doi.org/10.1149/1.1613668.
Bintang HM, Lee S, Shin S, Kim BG, Jung HG, Whang D, Lim HD. Stabilization effect of solid-electrolyte interphase by electrolyte engineering for advanced Li-ion batteries. Chem Eng J. 2021;424:130524. https://doi.org/10.1016/j.cej.2021.130524.
Zhu C, Zhang Y, Wu Z, Ma Z, Guo X, Guo F, Zhang J, Li Y. Closely packed Si@C and Sn@C nano-particles anchored by reduced graphene oxide sheet boosting anode performance of lithium ion batteries. J Mater Sci Technol. 2021;87:18. https://doi.org/10.1016/j.jmst.2020.12.075.
Lee BS, Yang HS, Lee KH, Han S, Yu WR. Rational design of a Si-Sn-C ternary anode having exceptional rate performance. Energy Storage Mater. 2019;17:62. https://doi.org/10.1016/j.ensm.2018.08.001.
Yang D, Shi J, Shi J, Yang H. Simple synthesis of Si/Sn@C-G anodes with enhanced electrochemical properties for Li-ion batteries. Electrochim Acta. 2018;259:1081. https://doi.org/10.1016/j.electacta.2017.10.117.
Hao Q, Hou J, Ye J, Yang H, Du J, Xu C. Hierarchical macroporous Si/Sn composite: easy preparation and optimized performances towards lithium storage. Electrochim Acta. 2019;306:427. https://doi.org/10.1016/j.electacta.2019.03.163.
Ma B, Luo J, Deng X, Wu Z, Luo Z, Wang X, Wang Y. Hollow silicon–tin nanospheres encapsulated by N-doped carbon as anode materials for lithium-ion batteries. ACS Appl Nano Mater. 2018;1(12):6989. https://doi.org/10.1021/acsanm.8b01793.
Morishita T, Hirabayashi T, Okuni T, Ota N, Inagaki M. Preparation of carbon-coated Sn powders and their loading onto graphite flakes for lithium ion secondary battery. J Power Sour. 2006;160(1):638. https://doi.org/10.1016/j.jpowsour.2006.01.087.
Lee BS, Yang HS, Lee KH, Han S, Yu WR. Rational design of a Si-Sn-C ternary anode having exceptional rate performance. Energy Storage Mater. 2018;17:62. https://doi.org/10.1016/j.ensm.2018.08.001.
Zhou ZW, Liu YT, Xie XM, Ye XY. Constructing novel Si@SnO2 core-shell heterostructures by facile self-assembly of SnO2 nanowires on silicon hollow nanospheres for large, reversible lithium storage. ACS Appl Mater Interfaces. 2016;8(11):7092. https://doi.org/10.1021/acsami.6b00107.
Ma T, Yu X, Li H, Zhang W, Cheng X, Zhu W, Qiu X. High volumetric capacity of hollow structured SnO2@Si nanospheres for lithium-ion batteries. Nano Lett. 2017;17(6):3959. https://doi.org/10.1021/acs.nanolett.7b01674.
Sui X, Huang X, Wu Y, Ren R, Pu H, Chang J, Zhou G, Mao S, Chen J. Organometallic precursor-derived SnO2/Sn-reduced graphene oxide sandwiched nanocomposite anode with superior lithium storage capacity. ACS Appl Mater Interfaces. 2018;10(31):26170. https://doi.org/10.1021/acsami.8b04851.
Ge M, Rong J, Fang X, Zhang A, Lu Y, Zhou C. Scalable preparation of porous silicon nanoparticles and their application for lithium-ion battery anodes. Nano Res. 2013;6(3):174. https://doi.org/10.1007/s12274-013-0293-y.
Ke CZ, Liu F, Zheng ZM, Zhang HH, Cai MT, Li M, Yan QZ, Chen HX, Zhang QB. Boosting lithium storage performance of Si nanoparticles via thin carbon and nitrogen/phosphorus co-doped two-dimensional carbon sheet dual encapsulation. Rare Met. 2021;40(6):1347. https://doi.org/10.1007/s12598-021-01716-1.
Song X, Wang Z, Liu Y, Wang C, Li L. A highly sensitive ethanol sensor based on mesoporous ZnO-SnO2 nanofibers. Nanotechnology. 2009;20(7):075501. https://doi.org/10.1088/0957-4484/20/7/075501.
Wang Q, Li H, Chen L, Huang XJ. Monodispersed hard carbon spherules with uniform nanopores. Carbon. 2001;39(14):2211. https://doi.org/10.1016/S0008-6223(01)00040-9.
Chen Y, Mao Q, Bao L, Yang T, Lu X, Du N, Zhang Y, Ji Z. Rational design of coaxial MWCNTs@Si/SiOx@C nanocomposites as extending-life anode materials for lithium-ion batteries. Ceram Int. 2018;44(14):16660. https://doi.org/10.1016/j.ceramint.2018.06.093.
Li B, Lai C, Zeng G, Qin L, Yi H, Huang D, Zhou C, Liu X, Cheng M, Xu P, Zhang C, Huang F, Liu S. Facile hydrothermal synthesis of Z-scheme Bi2Fe4O9/Bi2WO6 heterojunction photocatalyst with enhanced visible light photocatalytic activity. ACS Appl Mater Interfaces. 2018;10(22):18824. https://doi.org/10.1021/acsami.8b06128.
Kumar M, Oyedun AO, Kumar A. A review on the current status of various hydrothermal technologies on biomass feedstock. Renew Sust Energy Rev. 2018;81:1742. https://doi.org/10.1016/j.rser.2017.05.270.
Lou XW, Wang Y, Yuan C, Lee JY, Archer LA. Template-free synthesis of SnO2 hollow nanostructures with high lithium storage capacity. Adv Mater. 2006;18(17):2325. https://doi.org/10.1002/adma.200600733.
Gu Z, Li W, Chen Y, Xia X, Liu H. Synthesis of the microspherical structure of ternary SiOx@SnO2@C by a hydrothermal method as the anode for high-performance lithium-ion batteries. Sustain Energ Fuels. 2020;4(5):2333. https://doi.org/10.1039/d0se00053a.
Ao L, Wu C, Wang X, Xu Y, Jiang K, Shang L, Li Y, Zhang J, Hu Z, Chu J. Superior and reversible lithium storage of SnO2/graphene composites by silicon doping and carbon sealing. ACS Appl Mater Inter. 2020;12(18):20824. https://doi.org/10.1021/acsami.0c00073.
Dimov N, Kugino S, Yoshio M. Mixed silicon-graphite composites as anode material for lithium ion batteries. J Power Sour. 2004;136(1):108. https://doi.org/10.1016/j.jpowsour.2004.05.012.
Zuo P, Yin G, Ma Y. Electrochemical stability of silicon/carbon composite anode for lithium ion batteries. Electrochim Acta. 2007;52(15):4878. https://doi.org/10.1016/j.electacta.2006.12.061.
Qin J, Wu M, Feng T, Chen C, Tu C, Li X, Duan C, Xia D, Wang D. High rate capability and long cycling life of graphene-coated silicon composite anodes for lithium ion batteries. Electrochim Acta. 2017;256:259. https://doi.org/10.1016/j.electacta.2017.10.022.
Zhao Y, Wang LP, Xi S, Du Y, Yao Q, Guan L, Xu ZJ. Encapsulating porous SnO2 into a hybrid nanocarbon matrix for long lifetime Li storage. J Mater Chem A. 2017;5(48):25609. https://doi.org/10.1039/c7ta09544a.
Zhang T, Gao J, Fu LJ, Yang LC, Wu YP, Wu HQ. Natural graphite coated by Si nanoparticles as anode materials for lithium ion batteries. J Mater Chem. 2007;17(13):1321. https://doi.org/10.1039/b612967f.
Liang T, Hu R, Zhang H, Zhang H, Wang H, Ouyang Y, Liu J, Yang L, Zhu M. A scalable ternary SnO2-Co-C composite as a high initial coulombic efficiency, large capacity and long lifetime anode for lithium ion batteries. J Mater Chem A. 2018;6(16):7206. https://doi.org/10.1039/c8ta00957k.
Ngo DT, Le HTT, Pham XM, Jung JW, Vu NH, Fisher JG, Im WB, Kim ID, Park CJ. Highly porous coral-like silicon particles synthesized by an ultra-simple thermal-reduction method. J Mater Chem A. 2018;6(6):2834. https://doi.org/10.1039/c7ta09042k.
Xing Y, Shen T, Guo T, Wang X, Xia X, Gu C, Tu J. A novel durable double-conductive core-shell structure applying to the synthesis of silicon anode for lithium ion batteries. J Power Sour. 2018;384:207. https://doi.org/10.1016/j.jpowsour.2018.02.051.
Yong Y, Fan LZ. Silicon/carbon nanocomposites used as anode materials for lithium-ion batteries. Ionics. 2013;19(11):1545. https://doi.org/10.1007/s11581-013-0886-1.
Zhang Y, Zhao Y, Niu Y, Ren J, Hou H. Facile hydrothermal synthesis of double shelled Si@SnO2@C as advanced cathode for high-temperature lithium batteries. J Alloys Compd. 2021. https://doi.org/10.1016/j.jallcom.2020.157661.
Pan B, Peng X, Wang Y, An Q, Zhang X, Zhang Y, Teets TS, Zeng MH. Tracking the pyrolysis process of a 3-MeOsalophen-ligand based Co complex for promoted oxygen evolution reaction. Chem Sci. 2019;10(17):4560. https://doi.org/10.1039/c9sc00505f.
Yang S, Song H, Chen X. Electrochemical performance of expanded mesocarbon microbeads as anode material for lithium-ion batteries. Electrochem Commun. 2006;8(1):137. https://doi.org/10.1016/j.elecom.2005.10.035.
Zhang FZ, Ma YY, Jiang MM, Luo W, Yang JP. Boron heteroatom-doped silicon-carbon peanut-like composites enables long life lithium-ion batteries. Rare Met. 2022;41(4):1276. https://doi.org/10.1007/s12598-021-01741-0.
Acknowledgements
This work was financially supported by GRINM Science and Technology Innovation Fund (Nos. 2020DY0109 and 57222001), the Opening Project Fund of Materials Service Safety Assessment Facilities (No. MSAF-2021-001) and Guangdong High Level Innovation Research Institute (No. 2021B0909050001)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that they have no conflict of interest.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lei, Y., Li, S., Du, M. et al. Preparation of double-shell Si@SnO2@C nanocomposite as anode for lithium-ion batteries by hydrothermal method. Rare Met. 42, 2972–2981 (2023). https://doi.org/10.1007/s12598-023-02325-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12598-023-02325-w