
Abstract
Shellfish are recognized as a potential vehicle of viral disease and despite the control measures for shellfish safety there is periodic emergence of viral outbreaks associated with shellfish consumption. In this study a total of 81 mussel samples from Ría do Burgo, A Coruña (NW Spain) were analysed. Samples were collected in seven different harvesting areas with the aim to establish a correlation between the prevalence of norovirus (NoV) and hepatitis A virus (HAV) in mussel samples and the water quality. In addition, the genogroup of the detected HAV and NoV strains was also determined. The HAV presence was detected in 18.5 % of the samples. Contamination levels for this virus ranged from 1.1 × 102 to 4.1 × 106 RNA copies/g digestive tissue. NoV were detected in 49.4 % of the cases reaching contamination levels from 5.9 × 103 to 1.6 × 109 RNA copies/g digestive tissue for NoV GI and from 6.1 × 103 to 5.4 × 106 RNA copies/g digestive tissue for NoV GII. The χ²-test showed no statistical correlation between the number of positive samples and the classification of molluscan harvesting area based on the E. coli number. All the detected HAV strains belong to genogroup IB. NoV strains were assigned to genotype I.4, II.4 and II.6.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sewage pollution can contaminate shellfish growing-waters with various enteric viruses of human origin (Lees 2000). Epidemiological evidence suggests that these viruses are the most common pathogens transmitted by bivalve shellfish (Lees 2000; Le Guyader et al. 2000), although only norovirus (NoV) and hepatitis A virus (HAV) have been clearly implicated in outbreaks linked to shellfish consumption (Koopmans and Duizer 2004; Le Guyader et al. 2010; Webby et al. 2007). NoV is the most common pathogen causing gastroenteritis worldwide. The World Health Organization (WHO) (2008) estimates that there are 4.6 billion episodes of diarrhoeal illness with ~2.2 million associated deaths each year, largely attributable to contaminated food and drinking water in developing countries. In addition, acute gastroenteritis causes significant morbidity, and frequently requires hospitalization in developed countries (Wardlaw et al. 2010). Hepatitis A is the most serious viral infection linked to shellfish consumption causing a serious debilitating disease and even, occasionally, death (Lees 2000).
In an effort to control shellfish-borne infections, the EU has proposed several control measures for shellfish safety, which culminated in the implementation of Regulations 853/2004, 854/2004 and 1021/2008 (Anonymous 2004a, b, 2008). These standards rely exclusively on Escherichia coli numbers, which are routinely used to test the microbiological quality of shellfish and to classify shellfish harvesting areas. Despite all the efforts, periodic emergence of outbreaks associated with shellfish consumption with a presumed viral etiology continue to pose a real public health dilemma that results in substantial economic losses by the seafood industry and a lack of public confidence over shellfish (Romalde et al. 2002).
HAV is a non-enveloped 7.5 kb positive-stranded RNA virus that belongs to the Picornaviridae family, being the only member of genus Hepatovirus (Melnick 1992). Only one serotype of HAV has been identified worldwide. Despite this low amino acidic heterogeneity, there is substantial sequence variability allowing to classify HAV strains into seven different genotypes designed I–VII. Genotypes I and III have been further divided into subgenotypes A and B (Robertson et al. 1992).
NoV, classified in the family Caliciviridae, are a group of non-enveloped, icosahedral viruses, with a 7.5–7.7 kb positive-sense single-stranded RNA genome. NoV are genetically diverse, being classified into 29 genetic clusters within 6 genogroups (GI–GVI). Genogroups I and II strains most commonly affect humans (Ando et al. 2000; Wang et al. 1994).
The aim of this study was to determine the prevalence of HAV and NoV in mussel samples from Ría do Burgo, A Coruña (NW Spain) and its correlation with classification of molluscan harvesting area based on E. coli number. Sequence analysis of the detected strains was also carried out to know what HAV and NoV genogroups were circulating in this region.
Materials and Methods
Sample Collection and Processing
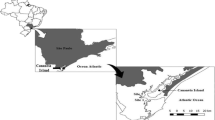
The current European legislation (Anonymous 2004a, b, 2008) classified the molluscan shellfish harvesting areas into 3 different quality levels, based in the E. coli number per gram of shellfish, as: A (<230 E. coli colony forming units [cfu]/100 g shellfish), B (<4,600 E. coli cfu/100 g shellfish) and C (4,600–46,000 E. coli cfu/100 g shellfish). A total of 81 mussels samples were obtained over a 18-month period (October 2010–March 2012) from seven different harvesting areas in Ría do Burgo, A Coruña (Galicia, NW Spain). Five of these harvesting areas are classified as B and two are classified as C (Fig. 1) by the official authority of Galician Regional Government.

Shellfish harvesting areas in Ría do Burgo; 1 Point 1 (43 20 38.8 N/8 21 31.59E), 2 Point 2 (43 22 2.20 N/8 23 20.27E), 3 Point 3 (43 20 42.59 N/8 22 51.76E), 4 Point 4 (43 20 25.31 N/8 23 2.99), 5 point 5 (43 20 4.9 N/8 22 52.9 W), 6 Point 6 (43 19 54.5 N/8 22 40.28), 7 Point 7 (43 19 47.80 N/8 22 20.70E)
From each mussel sample, 10 individuals were cleaned and separated from their valves in sterile conditions. The hepatopancreas were dissected and homogenized with a volume of 0.1 % peptone water pH 7.5 using a blender for 60 s. The homogenate was centrifuged 1,000×g for 5 min to recover the supernatant. RNA extraction of each molluscan sample was carried out in duplicate with commercial NucleoSpin® RNA Virus kit (Macherey-Nagel, Germany) following manufacturer’ specifications.
Prior to the RNA extraction, a known amount of mengovirus clone vMC0 was spiked into sample homogenates (10 μl of mengovirus stock, 103 pfu) to be employed as a control for the process of nucleic acid extraction.
Hepatitis A and Norovirus Detection and Quantification
Extracted RNA was used for HAV and NoV GI and GII detection by real-time RT-PCR (rtRT-PCR) with TaqMan probe. Primers and probe are shown in Table 1 (Costafreda et al. 2006; da Silva et al. 2007). Both HAV and NoV rtRT-PCRs were performed with platinum Quantitative RT-PCR Thermoscript One-Step System Kit (Invitrogen, France), following the manufacture’s recommendations. Amplification conditions were: reverse transcription at 55 °C for 1 h, denaturation at 95 °C for 5 min, followed by 45 cycles of amplification with a denaturation at 95 °C for 15 s, annealing at 60 °C for 1 min, and extension at 65 °C for 1 min. Viral RNA extracted from each sample was tested undiluted and at a tenfold dilution to reduce the effect of potential RT-PCR inhibitors.
Mengovirus were detected by rtRT-PCR with TaqMan probe under the same conditions employed for HAV and NoVs GI and GII detection. Primers and probe sequences are shown in Table 1. Extraction efficiency values were evaluated by comparing the Ct value for the vMC0-positive amplification control with the Ct value for mengovirus amplification in each examined sample. These results were classified as poor (<1 %), fair (1–10 %), or good (>10 %) (da Silva et al. 2007).
To test for the presence of RT-PCR inhibitors and to calculate rRT-PCR efficiency in both dilution of each examined sample, amplification using 2.5 μl of each extracted RNA and 2.5 μl of internal controls containing 103 genome copies of the respective virus type were evaluated in separate experiments (Costafreda et al. 2006; Lees 2010). The rtRT-PCR efficiency was calculated by comparing the Ct value of a sample mixed with internal controls to the Ct value of the internal control alone. Efficiency values were classified in the same three categories as the extraction efficiency (poor, fair and good) (da Silva et al. 2007).
In the samples with fair or good extraction efficiency and rtRT-PCR efficiency, the number of viral RNA copies/g tissue was estimated using standard curves generated from RNA transcripts. Quantification results were analysed by applying the corrective factors derived from virus RNA extraction efficiencies and the efficiencies of rtRT-PCR amplification. Only the samples with Ct values lower than 41 cycles were considered as positive. The limit of quantification of the method is 100 copies/g tissue.
Negative control well, containing nuclease free water and the rtRT-PCR mix for NoV GI and GII, were included on each assay.
Statistical Analysis
The correlation between the number of HAV-positive samples and the classification of molluscan harvesting areas based on E. coli numbers were determined. An analysis of the two-by-two contingence tables with positive and non-detected samples results from harvesting areas B and C, using the χ²-test (software Chidos for Windows) (Rossi 2002) under a predetermined alpha level of significance (0.05) and 1 degree of freedom were performed. In the same way, NoV GI, GII and GI/GII contamination were also analysed in separated assays.
Hepatitis A Virus and Norovirus Typing
To genotype the detected HAV strains, VP1-P2A and VP3-VP1 junction regions were sequenced. In brief, purified RNA of those positive samples for HAV detection were used to generate a cDNA with M-MLV Reverse transcriptase (Invitrogen, France). Complementary DNA was synthesized with 5 μl of purified RNA at 48 °C for 45 min, using reverse primer BR9 for VP1-P2A and HAV2 for VP3-VP1 (Table 1).
To amplify the VP1-P2A junction region two rounds of PCR were subsequently performed with Immolase DNA polymerase (Bioline, Germany). For the first round outer reverse BR9 and forward BR5 primers were used (Table 1). For the second round a seminested PCR was carried out with outer forward BR5 primer and inner reverse BR6 primer. Obtained seminested RT-PCR fragment size was 267 bp. To amplify the VP3-VP1 the same strategy was followed using the primers HAV1 and HAV 2 for the first round of PCR and HAV 2 and HAV 3 for the second one (Table 1) as previously described by Manso et al. (2010).
Genotyping of the detected NoV strains was carried out by sequencing a partial capsid gene region. For this purpose a semi-nested RT-PCR protocol with specific primers for NoV GI and NoV GII was performed. RT was carried out with the expand reverse transcriptase (Roche, Germany) and 5 μl of purified RNA and 100 μM of random hexamer stock as reverse primer following manufacturer’s specifications. The first round PCR was conducted using primers GOG1F and G1SKR for NoV GI and GOG2F and G2SKR for NoV GII. The second PCR for NoV GI was conducted with primers G1SKF and G1SKR. For NoV GII, the second round of PCR was carried out with primers G2SKF and G2SKR (Kageyama et al. 2003; Kojima et al. 2002; Pérez-Sautu et al. 2012) (Table 1). All the PCRs were conducted using expand high fidelity PCR system (Roche, Germany) following manufacturer’s specifications. RT-PCR amplicons for NoV GI and GII were 330 and 344 bp, respectively. Amplification products were analyzed by electrophoresis in 1.5 % agarose gels and visualized with etidium bromide. Amplicons were purified from the gel with QIAQuick Gel Extraction Kit (Qiagen) and sequenced.
Phylogenetic Analysis of HAV and NoV GI and GII Strains
Sequence analysis was performed with the DNASTAR Lasergene SEQMAN program. Phylogenetic reconstructions were performed based on HAV VP1-P2A and VP3-VP1 junction regions and for NoV partial capsid. Sequences of HAV and NoV GI and GII reference strains were retrieved from Genbank. Neighbour-joining (NJ) phylogenetic trees were performed with MEGA5 software using the Kimura-2-parameters and the heuristic search option respectively, and a bootstrap of 1,000 replicates in both cases (Balboa et al. 2011).
Results
HAV and NoV Detection
Of the 81 mussel samples collected from Ría do Burgo, 15 (18.5 %) were positive for the HAV presence. In the case of NoV, their presence was detected in 41 (49.4 %) of the analysed samples. In 7 samples (8.6 %) NoV were detected with primers designed for GI detection, while in 25 samples (30.8 %) NoV were detected with GII primers, and in 9 samples (11.1 %) NoV were detected with both GI and GII primers. Contamination levels for HAV ranged from 1.1 × 102 to 4.1 × 106 RNA copies/g digestive tissue; for NoV GI, from 5.9 × 103 to 1.6 × 109 RNA copies/g digestive tissue; and for NoV GII from 6.1 × 103 to 5.4 × 106 RNA copies/g digestive tissue. All the positive samples also yield high extraction efficiencies (>10 %), except 1 sample for HAV, 2 for NoV GI and 6 for NoV GII that yield fair efficiencies (1–10 %). In the samples in which HAV or NoV were not detected, extraction efficiencies yield high values in 20 of them, whereas the other five samples yield fair efficiencies. All the positive samples yielded high rtRT-PCR efficiencies (>10 %), except 6 samples detected for NoV GI that had rtRT-PCR efficiencies considered as fair (1–10 %).
Fifty-three samples were collected from harvesting areas classified as zone B. Of these samples, 10 (18.8 %) were positive for the HAV presence, whereas in 25 (47.1 %) the NoV presence was detected. Four (7.5 %) samples were positive for GI, 15 (28.3 %) were positive for GII and 6 were positive for both GI and GII. The other 28 samples analysed were collected in harvesting areas classified as zone C. HAV were detected in 5 (17.8 %) of them and NoV in 16 (57.1 %):3 (10.7 %) were positive for GI, 10 (35.7 %) were positive for GII and in 3 both the NoV GI and GII presence were detected. The percentages of HAV and NoV positive samples detected at each collection point as well as the contamination levels are shown in Table 2.
Seasonal distribution of the NoV presence in molluscan shellfish was observed. In harvesting areas classified as B, the percentage of positive samples during the cold months from October 2010 to March 2011 and from October 2011 to March 2011 were 73.4 and 63.4 %, respectively, whereas in the warm months from April 2011 to September 2011 was 37.5 %. In harvesting areas classified as C, the percentage of positive samples during the cold months from October 2010 to March 2011 and from October 2011 to March 2011 were 66.6 and 91.6 %, respectively, whereas in the warm months from April 2011 to September 2011 was 28.5 %.
As there is only one sample collected from point two, it was not considered to determine the correlation between HAV and NoV prevalence and the current classification of the molluscan harvesting area. The highest number of HAV-positive samples was detected in collection point 3 (B). In the case of NoV, GI detection rate was the highest at point 7 (C), GII in point 4 (B) and the confection of genogroups (GI/GII) in point 1(B). In all the cases contamination levels were higher in zones classified as B. However, the χ² test showed no statistical correlation between the number of positive samples and the classification of molluscan harvesting areas.
HAV and NoV Phylogeny
Visual inspection of VP1-P2A/VP3-VP1 tree built using the NJ algorithm with the 15 strains of HAV revealed that all the strains constitute a robust monophyletic group (bootstrap value of 99 %) belonging to subgenotype IB (Fig. 2). The similarity percentage within the VP1-P2A/VP3-VP1 concatenated sequences ranges from 99.1 to 100 %. Similarity percentage between isolated HAV strains and IB reference strain (HM-175) range from 98.2 to 99.1 %.

Phylogenetic reconstruction based on the concatenated VP1-P2A and VP3-VP1 junction region sequences of HAV and reference strains by neighbour-joining method. Bar, expected nucleotide substitutions per site. Only bootstrap values above 70 % are shown (1,000 re-samplings) at each branch point
Only in 39 % of the detected NoV strains, suitable sequences for subsequent phylogenetic analysis were obtained. A phylogenetic tree for each NoV genogroup was constructed. Genogroup I phylogenetic tree shows that all the analysed sequences belong to genotype I.4, constituting a robust monophyletic group (bootstrap value of 100 %), although showing some intraspecific diversity (Fig. 3). All strains shared 99.4–100 % of mutual similarity percentage and showed a similarity percentage with GI.4 reference strains ranging from 91.7 to 93.6 %. A phylogenetic tree for genogroup II strains shows that 8 of them represent a robust monophyletic group (bootstrap value of 100 %) within the genotype GII.4. Of the 8 NoV GII strains detected in shellfish from the Ría do Burgo, 7 were classified as GII.4 2006b variant, and the other one was GII.4 2010 variant. Sequences had a mutual similarity percentage ranging from 97.4 to 100 %. Similarity percentage between these strains and the reference strain for this genotype range from 93.4 to 94.3 %. The other two GII strains constitute a robust monophyletic group (bootstrap value of 85 %) belonging to genotype II.6 (Fig. 4). Similarity percentage within the cluster is 98 and 86.2 % among them and the reference strain for this genotype.

Phylogenetic reconstruction based on the partial capsid gene sequences of NoV GI strains and reference strains by neighbour-joining method. Bar, expected nucleotide substitutions per site. Only bootstrap values above 70 % are shown (1,000 re-samplings) at each branch point

Phylogenetic reconstruction based on the partial capsid gene sequences of NoV GII strains and reference strains by neighbour-joining method. Bar, expected nucleotide substitutions per site. Only bootstrap values above 70 % are shown (1,000 re-samplings) at each branch point
In the case of the 6 GI sequences, 50 % of the strains were previously detected with GII primers, 33.3 % were detected with both GI and GII primers pairs and 16.6 % with GI primers. Of the 10 NoV GII sequences, 50 % of the strains were detected using both GI and GII primers, 30 % with GII primers pair and 20 % with GI primer.
Discussion
With the aim to determine the correlation between the presence of HAV and NoV in mussels and the classification of seven different harvesting areas from Ría do Burgo, Galicia (NW Spain), a sensitive method of rtRT-PCR was used to evaluate the viral levels in the samples from the different harvesting areas. To decrease the inhibitor and false positive problem inherent to the use of PCR the analysis was made under the recommendations of the CEN group (Lees 2010). The use of mengovirus strain vMC0 has been demonstrated as a good external control for extraction efficiency due to its structural characteristics and its absence in environmental samples. Concentrations expressed for this virus correctly reflect the amount of virus present in the contaminated samples. It is important to take into account that molecular techniques are not able to infer viral infectivity, because viral genomes of non-infectious viruses are also detected. In spite of this limitation, molecular methods like PCR are considering as the most suitable tool to detect the presence of viruses in bivalve shellfish (Romalde et al. 2002). Result obtained from mussels collected in Ría do Burgo showed that NoV is more frequently detected than HAV (49.38 and 18.51 % of the samples, respectively). These data are in agreement with those obtained by other authors (Mesquita et al. 2011; Suffredini et al. 2012), although HAV detection rates are lower than those reported in bivalve shellfish from others geographical regions near to Ría do Burgo (Manso et al. 2010; Mesquita et al. 2011). The detection rate for HAV, NoV GII and contamination of both NoV GI/GII in the same sample is higher in harvesting areas classified as B, whereas for NoV GI the higher detection rate is found in zone C. In addition, the detected numbers of RNA copies/g digestive tissue yield larger values in samples from harvesting areas classified as B, both for NoV and HAV. However, χ²-statistical test showed that the number of positive samples for each virus and the classification of the harvesting areas based on the E. coli number are independent variables. These results support previous studies affirming that, at least in short-term studies, there is a lack of association between the actual assessment classification system of molluscan harvesting areas and the presence of human enteric viruses (Le Guyader et al. 2000, 2006, 2008; Romalde et al. 2002; Myrmel et al. 2004; Croci et al. 2007; Svraka et al. 2007; Vilariño et al. 2009). This fact and the inefficacy of the depuration to eliminate or reduce viral loads in shellfish (Baggi et al. 2001; Kingsley and Richards 2003), implicate shellfish as a potential public health risk.
According with previous studies, a season distribution in NoV detection in shellfish samples was observed, with the highest prevalence during the winter months (Formiga-Cruz et al. 2002; Flannery et al. 2009). It had been suggested that this seasonality could be attributed to several factors, including increased stability of viruses at a low water temperature, reduced solar inactivation and higher turbidity (Allwood et al. 2005; Cannon et al. 2006).
Phylogenetic analysis of HAV showed that all the strains belong to subgenotype IB. These results are in agreement with previous studies suggesting a high prevalence of genotype I worldwide (Rodrigues et al. 2007), although in Europe a co-circulation of subgenotypes IA and IB exists, with the first one more common (Cristina and Costa-Mattioli 2007). The presence of subgenotype IB in the samples together with previous results obtained from shellfish collected in the Ría de Vigo, Galicia (Manso et al. 2010) and in Portugal (Mesquita et al. 2011) suggest a possible endemic circulation on the west coast of the Iberian Peninsula.
It is well-recognised that NoV strains diversity is extremely high. For this reason, separate assays for detection of both genogroups are recommended (Lees 2010) using a target region for the amplification the 5′ end of the NoV capsid sequence. It is a relatively conserved region in NoV genome used to amplify a wide range of NoV GI and GII genotypes with a conserved set of primers (Katayama et al. 2002). Nevertheless, analysis of NoV genotype revealed that the strain genotype does not always correlates with the results of detection with primers pairs GI and/or GII. Although the target genome region for NoV detection is relatively conserved, a variety of single nucleotide polymorphisms (SNPs) are found within this region making the suitable primer and probe design difficult (Kageyama et al. 2003; Loisy et al. 2005; Trujillo et al. 2006). Chosen NoV GI and GII primers and probe set are specific for NoV detection, but genotype assignment must be confirmed by sequences analysis.
The high incidence of NoV infections seems to be related to the emergence of new GII.4 variants that had evolved by genetic drift of the capsid gene (Bull et al. 2010). It has been shown for the first time that GII.4 NoV variant US95/96 was predominant worldwide during the winter of 1995–1996 (Vinjé et al. 1997). In 2002, a new variant named Farmington Hills was detected and became predominant (Widdowson et al. 2004). Since 2002, a new GII.4 variant has become predominant every 2 or 3 years. Strains detected in mussels from Ría do Burgo belonged to genotypes I.4, II.4 and II.6, with genotype II.4 the most common. Within this genotype, detected strains belonged to variants 2006b and 2010. This fact reflects the situation in the human population of this geographical region, in which 2006 variant has been displaced by the 2010 variant during the year 2011 (unpublished data). In another analysis carried out by Mesquita et al. (2011) in Portugal, all the NoV strains detected in shellfish also belonged to genotype II.4. This NoV genotype has been predominant on a global scale for the past years (Kanerva et al. 2009; Le Guyader et al. 2010). Molecular epidemiological studies show an apparent worldwide dominance in NoV GII over GI strains in fecal samples (Victoria et al. 2009) suggesting that the higher GII viral load could facilitate the transmission of the virus. However, it is important to note that it has been reported that NoV GI is more often implicated in outbreaks because of its higher resistance to breakdown during wastewater treatment (Le Guyader et al. 2006; da Silva et al. 2007).
From the results obtained, it seems clear that virological analyses must be included in any sanitary survey for classification of shellfish harvesting areas, and that the shift of NoV variants that occurred over the last years in clinical strains has a rapid reflection in the prevalence of viral variants in shellfish.
References
Allwood, P. B., Malik, Y. S., Maherchandani, S., Hedberg, C. W., & Goyal, S. M. (2005). Effect of temperature on the survival of F-specific RNA coliphage, feline calicivirus, and Escherichia coli in chlorinated water. International Journal of Environmental Research and Public Health, 2(3–4), 442–446.
Ando, T., Noel, J. S., & Fankhauser, R. L. (2000). Genetic classification of ‘‘Norwalk-like viruses’’. Journal of Infectious Disease, 181(2), 336–348.
Anonymous. (2004a). Commission regulation (EC) No 853/2004 of 29 April 2004 laying down specific hygiene rules for food of animal origin. Official Journal, L139, 22–82.
Anonymous. (2004b). Commission regulation (EC) No 854/2004 of 29 April 2004 laying down specific rules for the organization of official controls on products of animal origin intended for human consumption. Official Journal, L139, 83–127.
Anonymous. (2008). Commission regulation (EC) No 1021/2008 of 17 October 2008 amending Annexes I, II and III to Regulation (EC) No 854/2004 laying down specific rules for the organization of official controls on products of animal origin intended for human consumption and Regulation (EC) No 2076/2005 as regards live bivalve molluscs, certain fishery products and staff assisting with official controls in slaughterhouses. Official Journal, L277, 15–17.
Baggi, F., Demarta, A., & Peduzzi, R. (2001). Persistence of viral pathogens and bacteriophages during sewage treatment: Lack of correlation with indicator bacteria. Research in Microbiology, 152(8), 743–751.
Balboa, S., Bermudez-Crespo, J., Gianzo, C., López, J. L., & Romalde, J. L. (2011). Proteomics and multilocus sequence analysis confirm intraspecific variability of Vibrio tapetis. FEMS Microbiology Letters, 324(1), 80–87.
Bull, R. A., Eden, J. S., Rawlinson, W. D., & White, P.A. (2010). Rapid evolution of pandemic noroviruses of the GII.4 lineage. PLOS Pathogens, 6(3), e1000831.
Cannon, J. L., Papafragkou, E., Park, G. W., Osborne, J., Jaykus, L. A., & Vinjé, J. (2006). Surrogates for the study of norovirus stability and inactivation in the environment: A comparison of murine norovirus and feline calicivirus. Journal of Food Protection, 69(11), 2761–2765.
Costafreda, M. I., Bosch, A., & Pintó, R. M. (2006). Development, evaluation, and standardization of a real-time TaqMan reverse transcription-PCR assay for quantification of hepatitis A virus in clinical and shellfish samples. Applied and Environmental Microbiology, 72(6), 3846–3855.
Cristina, J., & Costa-Mattioli, M. (2007). Genetic variability and molecular evolution of hepatitis A virus. Virus Research, 127(2), 151–157.
Croci, L., Nadia, M., Suffredini, E., Pavoni, E., Di Pasquale, S., Fallacara, F., et al. (2007). Assessment of human enteric viruses in shellfish from the northern Adriatic sea. International Journal of Food Microbiology, 114(2), 252–257.
Da Silva, A. K., Le Saux, J. C., Parnaudeau, S., Pommepuy, M., Eimelech, M., & Le Guyader, F. S. (2007). Evaluation of removal of norovirus during wastewater treatment, using realtime reverse transcription-PCR: Different behaviors of genogroups I and II. Applied and Environmental Microbiology, 73(24), 7891–7897.
Flannery, J., Keaveney, S., & Doré, W. (2009). Use of FRNA bacteriophages to indicate the risk of norovirus contamination in Irish oysters. Journal of Food Protection, 72(11), 2358–2362.
Formiga-Cruz, M., Tofiño-Quesada, G., Bofill-Mas, S., Lees, D. N., Henshilwood, K., et al. (2002). Distribution of human virus contamination in shellfish from different growing areas in Greece, Spain, Sweden, and the United Kingdom. Applied and Environmental Microbiology, 68(12), 5990–5998.
Kageyama, T., Kojima, S., Shinohara, M., Uchida, K., Fukushi, S., Hoshino, F. B., et al. (2003). Broadly reactive and highly sensitive assay for Norwalk-like viruses based on real-time quantitative reverse transcription-PCR. Journal of Clinical Microbiology, 41(4), 1548–1557.
Kanerva, M., Maunula, L., Lappalainen, M., Mannonen, L., von Bonsdorff, C. H., & Anttila, V. J. (2009). Prolonged norovirus outbreak in a Finnish tertiary care hospital caused by GII.4-2006b subvariants. Journal of Hospital Infection, 71(3), 206–213.
Katayama, K., Shirato-Horikoshi, H., Kojima, S., Kageyama, T., Oka, T., Hoshino, F., Fukushi, S., Shinohara, M., et al. (2002). Phylogenetic analysis of the complete genome of 18 Norwalk-like viruses. Virology, 299(2), 225–239.
Kingsley, D. H., & Richards, G. P. (2003). Persistence of hepatitis A virus in oysters. Journal of Food Protection, 66(2), 331–334.
Kojima, S., Kageyama, T., Fukushi, S., Hoshino, F. B., Shinohara, M., Uchida, K., et al. (2002). Genogroup-specific PCR primers for detection of Norwalk-like viruses. Journal of Virological Methods, 100(1–2), 107–114.
Koopmans, M., & Duizer, E. (2004). Foodborne viruses: An emerging problem. International Journal of Food Microbiology, 90(1), 23–41.
Le Guyader, F. S., Bon, F., De Medici, D., Parnaudeau, S., Bertone, A., Crudeli, S., et al. (2006). Detection of multiple noroviruses associated with an international gastroenteritis outbreak linked to oyster consumption. Journal of Clinical Microbiology, 44(11), 3878–3882.
Le Guyader, F., Haugarreau, L., Miossec, L., Dubois, E., & Pommepuy, M. (2000). Three years study to asses human enteric virus in shellfish. Applied an Environmental Microbiology, 66(8), 3241–3248.
Le Guyader, F. S., Krol, J., Ambert-Balay, K., Ruvoen-Clouet, N., Desaubliaux, B., Parnaudeau, S., et al. (2010). Comprehensive analysis of a norovirus-associated gastroenteritis outbreak, from the environment to the consumer. Journal of Clinical Microbiology, 48(3), 915–920.
Le Guyader, F. S., Le Saux, J. C., Ambert-Balay, K., Krol, J., Serais, O., Parnaudeau, S., et al. (2008). Aichi virus, norovirus, astrovirus, enterovirus, and rotavirus involved in clinical cases from a French oyster-related gastroenteritis outbreak. Journal of Clinical Microbiology, 46(12), 4011–4017.
Lees, D. N. (2000). Viruses and bivalve shellfish. International Journal of Food Microbiology, 59, 81–116.
Lees, D. N. (2010). International standardisation of a method for detection of human pathogenic viruses in molluscan shellfish. Food and Environmental Virology, 2(3), 146–155.
Loisy, F., Atmar, R. L., Guillon, P., Le Cann, P., Pommepuy, M., & Le Guyader, F. S. (2005). Real-time RT-PCR for norovirus screening in shellfish. Journal of Virological Methods, 123(1), 1–7.
Manso, C. F., Polo, D., Vilariño, M. L., & Romalde, J. L. (2010). Genotyping of hepatitis A virus detected in bivalve shellfish in Galicia (NW Spain). Water Science and Technology, 61(1), 15–24.
Melnick, J. L. (1992). Properties and classification of hepatitis A virus. Vaccine, 10(Supp 1), 24–26.
Mesquita, J. R., Vaz, L., Cerqueira, S., Castilho, F., Santos, R., Monteiro, S., et al. (2011). Norovirus, hepatitis A virus and enterovirus presence in shellfish from high quality harvesting areas in Portugal. Food Microbiology, 28(5), 936–941.
Myrmel, M., Berg, E. M. M., Rimstad, E., & Grinde, B. (2004). Detection of enteric viruses in shellfish from the Norwegian coast. Applied and Environmental Microbiology, 70(5), 2678–2684.
Pérez-Sautu, U., Sano, D., Guix, S., Kasimir, G., Pintó, R. M., & Bosch, A. (2012). Human norovirus occurrence and diversity in the Llobregat river catchment, Spain. Environmental Microbiology, 14(2), 494–502.
Pintó, R. M., Costafreda, M. I., & Bosch, A. (2009). Risk assessment in shellfish-borne outbreaks of hepatitis A. Applied and Environmental Microbiology, 75(23), 7350–7355.
Robertson, B. H., Jansen, R. W., Khanna, B., Totsuka, A., Nainan, O. V., Siegl, G., et al. (1992). Genetic relatedness of hepatitis A virus strains recovered from different geographical regions. Journal of General Virology, 73(6), 1365–1377.
Rodrigues, L., Pista, A., Oliveira, I., Água-Doce, C., Manita, C., & Paixão, M. T. (2007). Molecular epidemiology of hepatitis A virus in a group of portuguese citizens living in Lisboa area. Journal of Medical Virology, 79(5), 483–487.
Romalde, J. L., Area, E., Sánchez, G., Ribao, C., Torrado, I., Abad, X., et al. (2002). Prevalence of enterovirus and hepatitis A virus in bivalve mollusks from Galicia (NW Spain): Inadequacy of the EU standards of microbiological quality. International Journal of Food Microbiology, 74(1–2), 119–130.
Rossi, G. (2002). Chidos software for Chi Square test. University of Verona. http://psico.univr.it/germano/soft/chiquadro.php.
Suffredini, E., Magnabosco, C., Civettini, M., Rossetti, E., Arcangeli, G., & Croci, L. (2012). Norovirus contamination in different shellfish species harvested in the same production areas. Journal of Applied Microbiology, 113(3), 686–692.
Svraka, S., Duizer, E., Vennema, H., de Bruin, E., van der Veer, B., Dorresteijn, B., et al. (2007). Etiological role of viruses in outbreaks of acute gastroenteritis in The Netherlands from 1994 through 2005. Journal of Clinical Microbiology, 45(5), 1389–1394.
Trujillo, A. A., McCaustland, K. A., Zheng, D. P., Hadley, L. A., Vaughn, G., Adams, S. M., et al. (2006). Use of TaqMan real-time reverse transcription-PCR for rapid detection, quantification, and typing of Norovirus. Journal of Clinical Microbiology, 44(4), 1405–1412.
Victoria, M., Guimarães, F., Fumian, T., Ferreira, F., Vieira, C., Leite, J. P., et al. (2009). Evaluation of an adsorption-elution method for detection of astrovirus and norovirus in environmental waters. Journal of Virological Methods, 156(1–2), 73–76.
Vilariño, M. L., Le Guyader, F. S., Polo, D., Schaeffer, J., Kröl, J., & Romalde, J. L. (2009). Assessment of human enteric viruses in cultured and wild bivalve molluscs. International Microbiology, 12(3), 145–151.
Vinjé, J., Altena, S. A., & Koopmans, M. P. (1997). The incidence and genetic variability of small round-structured viruses in outbreaks of gastroenteritis in The Netherlands. Journal of Infection Disease, 176(5), 1374–1378.
Wang, J., Jiang, X., Madore, H. P., Gray, J., Desselberger, U., Ando, T., et al. (1994). Sequence diversity of small, round-structured viruses in the Norwalk virus group. Journal of Virology, 68(9), 5982–5990.
Wardlaw, T., Salama, P., Brocklehurst, C., Chopra, M., & Mason, E. (2010). Diarrhoea: Why children are still dying and what can be done. The Lancet, 375(9718), 870–872.
Webby, R. J., Carville, K. S., Kirk, M. D., Greening, G., Ratcliff, R. M., Crerar, S. K., et al. (2007). International distributed frozen oyster meat causing multiple outbreaks of Norovirus infections in Australia. Clinical Infectious Diseases, 44(8), 1026–1031.
Widdowson, M. A., Cramer, E. H., Hadley, L., Bresee, J. S., Beard, R. S., Bulens, S. N., et al. (2004). Outbreaks of acute gastroenteritis on cruise ships and on land: Identification of a predominant circulating strain of norovirus-United States, 2002. Journal of Infectious Disease, 190(1), 27–36.
Yun, H., Kim, S., Soon Byun, K., Kwon, S. Y., Yim, H. J., Lim, Y., et al. (2008). Genetic analysis of HAV strains isolated from patients with acute Hepatitis in Korea, 2005–2006. Journal of Medical Virology, 80(5), 777–784.
Acknowledgment
This study was supported in part by grant 10MMA200010PR from the Consellería de Economía e Industria, Xunta de Galicia (Spain). C.F.M. acknowledges the Xunta the Galicia (Spain) for a research fellowship. We acknowledge INTECMAR (Instituto Tecnológico para o Contro do Medio Mariño, Xunta de Galicia) for kindly supplying shellfish samples.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Manso, C.F., Romalde, J.L. Detection and Characterization of Hepatitis A Virus and Norovirus in Mussels from Galicia (NW Spain). Food Environ Virol 5, 110–118 (2013). https://doi.org/10.1007/s12560-013-9108-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12560-013-9108-2