
Abstract
Bacteriophage T4 initially recognizes its host cells using its long tail fibers. Long tail fibers consist of a phage-proximal and a phage-distal rod, each around 80 nm long and attached to each other at a slight angle. The phage-proximal rod is formed by a homo-trimer of gene product 34 (gp34) and is attached to the phage-distal rod by a monomer of gp35. The phage-distal rod consists of two protein trimers: a trimer of gp36, attached to gp35, although most of the phage-distal rod, including the receptor-binding domain, is formed by a trimer of gp37. In this review, we discuss what is known about the detailed structure and function of the different long tail fiber domains. Partial crystal structures of gp34 and gp37 have revealed the presence of new protein folds, some of which are present in several repeats, while others are apparently unique. Gp38, a phage chaperone protein necessary for folding of gp37, is thought to act on an α-helical coiled-coil region in gp37. Future studies should reveal the remaining structure of the long tail fibers, how they assemble into a functional unit, and how the long tail fibers trigger the infection process after successful recognition of a suitable host bacterium.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the extracellular state bacteriophages, similar to all viruses, consist of at least a protein capsid surrounding a nucleic acid genome (Prasad and Schmid 2012). The Caudovirales, the tailed bacteriophages, have a double-stranded DNA containing icosahedral or prolate head (Ackermann 2003). One of the vertices of the head is unique and sports a tail, a protein structure that functions at the beginning of the infection process. Tails define Caudovirales families and may be short (Podoviridae), long and flexible (Siphoviridae), or medium in length, rigid and contractile—shortening to extend an inner tube through the outer layers of the bacterial cell to provide an entry tube for the genome—(Myoviridae). The tails of many of the Myoviridae have two sets of receptor binding proteins at the distal end of the tail (Goldberg et al. 1994). The short tail fibers are an integral part of the baseplate (the end of the tail) and trigger the tail’s contraction after binding to the host cell (Leiman et al. 2004; Taylor et al. 2016). Long tail fibers (LTFs) are also attached to the tail but act before the short tail fibers, binding to the host cell and orienting the phage so that the short tail fibers are deployed correctly (Crawford and Goldberg 1980; Goldberg et al. 1994). The binding of the long tail fibers may also support subsequent triggering of the baseplate.
For bacteriophage T4, a member of the Myoviridae family, many of the details of the infection process have been previously described (Wood and Crowther 1983; Goldberg et al. 1994; Leiman et al. 2004; Hu et al. 2015; Taylor et al. 2016). In the free state, the long tail fibers of T4 are folded upwards against the tail, neck, and head domains, although, under favorable conditions for infection, an LTF can be released from the neck and head and probe the surroundings for a suitable host receptor site (lipopolysaccharide and/or OmpC). If a potential site is found, two-dimensional diffusion of the phage on the cell surface occurs until a second, then a third and up to six LTFs bind to the cell surface. If three or more LTFs bind to the host, the encountered cell is very likely to be a suitable replication host, and a signal is transferred, possibly through differential sampling of the angles at which the LTF attaches to the baseplate. The baseplate then changes conformation from the hexagonal conformation to the star-conformation and the short tail fibers extend, releasing their carboxy-terminal ends to bind tightly to the core region of the host LPS. The outer tail sheath now contracts, driving the inner tail tube through the bacterial outer membrane and periplasm, allowing the end of the inner tube to interact with the bacterial inner membrane. Phage DNA is then ejected into the bacterial cytoplasm, safe from periplasmic nucleases and primed to direct the synthesis of new phage particles.
Structurally, the LTFs of bacteriophage T4 are unusual protein complexes. In their final assembled form, they consist of two ~ 80-nm rods, joined end-to-end at a fixed angle of around 160°. LTFs are only 3–5 nm in diameter. During infection, LTFs are assembled independently of the rest of the capsid and then attached to the end of the tail (Wood and Bishop 1973). Before being joined to the tail, the bent rods have a small knob at the end that will attach to the tail and taper to a thinner structure at the distal end (Ward et al. 1970). This thin end, usually described as the needle tip, contains the receptor binding sequences of the LTF. This was first shown by electron microscopy of T2 and T4 phages infecting Escherichia coli (Simon and Anderson 1967).
The low-resolution structure of the LTFs and their assembly have been extensively reviewed (Bishop et al. 1974; Wood and Crowther 1983; Goldberg et al. 1994; Leiman et al. 2004). The final assembled LTF is composed of a total of ten proteins, the products of four genes. The products of genes 34, 36, and 37 are each present as homo-trimers along with a monomeric copy of the gene 35 protein (Cerritelli et al. 1996). The product from gene 57 is a chaperone needed for correct assembly of gp34 and 37 trimers, but is not part of the final assembled complex. Additionally, the gene 38 protein is necessary for folding of the glycoprotein 37 (gp37) monomers and is also not part of the final LTF, although in many related phages it does remain attached to the end of the fiber and acts as the receptor-binding protein. The product of gene 63 improves the efficiency of attachment of the assembled tail fibers to the tail, but it is not absolutely required for attachment. A series of serological, genetic, and protein isolation experiments (see reviews cited in preceding text) demonstrated that the gene 34 and gene 37 products assemble independently into homo-trimeric gp34 and gp37 complexes, respectively. The gene 36 proteins assemble on the gp37 complex to form a gp36–gp37 complex. Then the gene 35 protein joins to the gp36 complex free end to form a gp35–gp36–gp37 complex that is usually described as the distal half fiber. The homo-trimer of gp34 is the proximal half fiber (proximal and distal are in relation to the final assembled LTF relative to the tail), and it attaches to the gp35–gp36–gp37 complex to form the complete LTF, which then can join to the tail with the aid of the gene 63 product.
The long, thin, final assembled structure shown in Fig. 1 also reflects a somewhat linear arrangement of the proteins within the LTF. That is, the proteins (probably except for the gene 35 protein) are arranged in trimers whose N-termini are together at one end of the assembly and whose C-termini are at the other end. Thus, the N-termini of the gp34 trimers attach to the tails while the C-termini bind to the gene 35 protein. Likewise, the N-termini of the gp36 trimers bind to gene 35 proteins while the C-termini of the gp36 complexes bind to the N-termini of gp37 trimers. Finally, the C-termini of gp37 trimers contain the receptor-binding amino acid (aa) sequences for binding to the host bacterium. These binding amino acid sequences at the termini are highly conserved between related phages, but amino acid sequences of the central regions (which presumably correspond to much of the length of the tail fiber, i.e., the rod regions) are not conserved (Riede et al. 1985; Hyman and Harrah 2014), which is somewhat surprising given that the tail fibers of related bacteriophages have the same basic assembled structure of long, thin, bent rods.

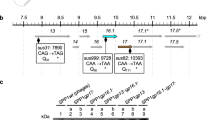
Domainal organization of the bacteriophage T4 long tail fiber. a Outline of the bacteriophage T4 long tail fiber (LTF), composed of gene products (gp)34–gp37. The proximal tail fiber is on the left and the distal tail fiber is on the right, as in the marked tail fiber in the whole phage drawing. Lines indicate approximate borders between the four proteins. b Domainal and protein functional correspondence for the LTF. Protein locations are indicated above the outline and the functional domains are indicated under the outline. Mass domain names are shown for the proximal half-fiber (P1–P5), the knee-cap (KC), and the distal half-fiber (D1–D11), and refer to the mass domains identified by Cerritelli et al. (1996). The correspondence between the functional and mass domains as well as the position of the mass domains is approximate. c Detailed positions of functional domains, with the numbers after each function description indicating the amino acid (aa) residues in that protein for that domain when known. Amino (N) and carboxy (C) ends of gp34, gp36, and gp37 are indicated. There is no corresponding information for gp35. The two deletions mentioned in the text in the central rod region of gp37 are also shown
The studies cited above led to the simple model of the long tail fiber structure shown in Fig. 1, with distinct functional domains needed for assembly. Because of the large size and simple linear structure of the assembled tail fiber, crystallizing whole tail fibers has proved challenging, although some fragments of tail fiber proteins have been assembled, crystallized, and their structures solved. For other domains, mutant and other analysis supports specific functional roles for particular domains. In this review, we discuss current knowledge on the different functional domains of this protein complex. In discussing these structures the word domain will be used in two distinct ways. Functional domains will be identified by descriptions detailed above and by specific amino acid residues when known from solved structures. However, a second use of the term domain comes from the work of Cerritelli et al. (1996) who studied the overall structure of the long tail fibers by electron microscopy. Using scanning transmission electron microscopy mass mapping and other techniques, they divided the long tail fiber into 17 mass domains. As shown in Fig. 1b, these include five in the proximal tail fiber [P1 (baseplate attachment)–P5 (gp35 binding)]; a single knee-cap domain (essentially the gp 35 protein joined to the proximal and distal tail fibers); and 11 distal tail fiber domains [D1 (gp36–gp35 binding)–D11 (gp37 needle tip end)]. These mass domains approximately correspond to thicker and thinner regions of the tail fiber and, as we now know from analysis of partial structures, correspond to varying changes in structural motifs. We will use the mass domain designations in the following sections as well.
The gp37 tip and collar domains
For crystallographic studies, a C-terminal fragment of a gp37 trimer containing domains D9, D10, and D11 was produced by co-expression with the two chaperone proteins gp57 and gp38 (Bartual et al. 2010a). Controlled proteolysis yielded a protein consisting of the D10 and D11 domains (residues of 811–1026 of the 1026-aa protein), which was crystallized and the structure solved (Bartual et al. 2010b).
The 20-nm-long structure (PDB ID 2XGF) clearly exhibited the D10 and D11 domains in the expected overall shape, as revealed by Cerritelli et al. (1996), while the detailed folds revealed some surprises (Fig. 2). The D10 domain is formed by amino acids 811–881 and 1010–1026. Each monomer contains a small β-sandwich of antiparallel sheets with an α-helix inserted into a loop of the outside β-sheet. The three inner sheets provide the trimer interactions. The same fold had been observed before in the T4 baseplate proteins gp10, gp11, and gp12 and has been named the collar domain (Leiman et al. 2000; van Raaij et al. 2001; Leiman et al. 2006).

Structure of the phage T4 LTF tip. a Side view of a ribbon representation of the gp37 trimer fragment containing the D10 and D11 domains. The three chains of the protein homo-trimer are in green, red, and blue, respectively. The iron ions are shown as orange balls, and the histidine side chains coordinating them are visualized. Residues forming the approximate domain boundaries are labeled. b End-on view of the receptor-binding head domain in which the interweaving of the three chains is clearly visible. Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081) (Pettersen et al. 2004)
Next to the collar domain is a small entwined region that completes the D10 domain. The C-terminus of each gp37 is folded back into the D10 domain, making the D11 needle domain an insertion into the D10 domain, projecting outwards. In gp12, a similar insertion is present, although not in the shape of a needle. Instead, the insertion has a globular shape and consists of intertwined strands and a single central metal ion (PDB ID 1OCY; Thomassen et al. 2003). The baseplate gp10 also has an insertion in the same place, which forms a 6-nm long shaft domain (PDB ID 2FKK; Leiman et al. 2006). In gp11, there is an insertion in a different loop, pointing to the side (PDB ID 1EL6; Leiman et al. 2000).
D11 is an elongated structure in which almost all amino acids are in an extended conformation. Residues 882–931 and 960–1009 form the needle domain. Inward-facing histidine residues coordinate seven iron ions in the needle domain, of which the ones coordinating Fe1, Fe5, and Fe7 are located in the forward strand and the ones coordinating Fe2, Fe3, Fe4, and Fe6 are in the reverse strand. These histidines are part of HXH motifs. At the end of the needle domain, and thus of the gp37 trimer and the LTF, is a small head domain formed by residues 932–959. Except for the head domain, the sequences of the D10 and D11 domain are conserved in other phages (like lambda), suggesting they have a structurally conserved end to their LTF, but with a different receptor-binding function. Interestingly, the lambda side tail fiber has two small inserts in the needle domain, one in the forward strand and another in the reverse strand, and the latter contains an extra HXH motif, suggesting it may bind eight iron ions instead of seven (Bartual et al. 2010b).
The coiled-coil α-helical segment: site of gp38 activity
The normal assembly process of gp37 trimers requires the products of both genes 57 and 38, neither of which are found in the assembled LTF. In 1976, Bishop and Wood (1976) described a suppressor mutation of gene 38 double amber mutations that could assemble LTFs normally, albeit at reduced efficiency, at 25 °C but not 42 °C. The suppressor mutation, designated ts3813, was later mapped to the carboxyl end of gene 37 (Snyder and Wood 1989). When sequenced, ts3813 was found to be a 21-bp duplication (Hashemolhosseini et al. 1994). Hashemolhosseini and colleagues proposed that this extended sequence substituted for the action of the gene 38 product in initiating gp37 trimer assembly. Edward Goldberg (personal communication) later noticed that the duplicated protein sequence had a coiled-coil α-helix motif, a motif that commonly aligns proteins in multimeric complexes (Branden and Tooze 1999). Cerritelli et al. (1996) had noted the potential of a coiled-coil α-helix motif in the wild-type gene 37 protein but did not connect this with the ts3813 extension. Goldberg’s observation suggested that the region, supported or stabilized by the gene 38 product, functioned to align the three copies of gp37 to begin forming the assembled gp37 portion of the distal tail fiber.
To test this possibility, Goldberg and colleagues (including one of this review’s authors, PH) constructed phage containing even longer duplications of the putative coiled-coil α-helix (Qu et al. 2004). These motifs consist of a minimal amino acid heptad which forms two turns of an α-helix. The wild-type gene 37 protein contains two heptads at amino acids 795–809, and the ts3813 mutant has three heptads. The engineered mutants had four and five heptads. As predicted, the four- and five-heptad mutants were able to grow at temperatures up to 50 °C without a functional gene 38 protein, supporting the hypothesis that the coiled-coil α-helix was the site of action of the gene 38 protein in gp37 assembly initiation.
One question suggested by this is why the phage would have maintained a requirement for the gene 38 protein rather than extending the coiled-coil region? There are several possibilities. First, direct DNA duplications are not stable in the bacteriophage T4 genome. During the work under discussion, spontaneous mutations that removed part of the duplication were identified. Bacteriophage T4 uses recombination as one mode of DNA replication initiation, so it is not surprising that duplications might be rapidly removed (Kreuzer and Morrical 1994). Second, longer exact duplicates of the coiled-coil α-helix may misalign, interfering with efficient assembly of the gp37 trimer. Third, longer coiled-coil α-helices may be too stable for efficient assembly. In the bacteriophage T4 gene 37 protein, the two coiled-coil α-helix heptads are found at amino acids 795–809. These amino acids are located just amino-terminal of the collar domain, between the wider rod region of gp37 and the collar domain and corresponding to the thin part between the D9 and D10 domains observed by Cerritelli et al. (1996). Duplications might extend this thin segment, perhaps making the fiber more sensitive to proteases or rendering it too flexible at this site.
The central part of the distal rod region
The structure of the central rod of the assembled gp37 has not been determined experimentally, but it is likely to function as a stiff rod. Between residues 88 and 104 there is a partial repetition of a repeat that is also present six times in gp12 and ten times in gp34 (Cerritelli et al. 1996; van Raaij et al. 2001; Taylor et al. 2016; see section The phage-proximal rod: gp34). As in the gp12 and gp34 complexes, this partial repeat likely forms a β-strand interacting with its homologs from the other two monomers. However, in gp12 and gp34, most of the repeats are longer, as described by Granell et al. (2017). The extra parts of these repeats are not conserved in gp37.
Residues 483–738 show homology to residues 968–1196 of gp34 and can be aligned with 28% identities and 42% similarities (BLAST-P). In gp34, these residues form the second part of the P3 domain, a stretch of triple β-helix, the P4 domain, another stretch of triple β-helix, and part of the P5 domain (Granell et al. 2017; see also section The phage-proximal rod: gp34). According to Cerritelli et al. (1996), residues 483–738 of gp37 are in the D7, D8, and D9 domains; it is therefore likely that the D7, D8, and D9 domains of the LTFs resemble their P3, P4, and P5 domains, including the triple β-helix regions that connect them. This, in turn, suggests that gp34 and gp37 may have resulted from a gene duplication event in the evolution of T4-like phages.
The gp36 assembly/binding sequences
The amino end of the gp37 protein trimer binds to the carboxyl end of the P36 trimer. The overall structure of the LTF does not show an obvious joint. The joint is not covalent since sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis of whole phage and of LTFs separates the proteins into distinct bands (King and Laemmli 1971; Earnshaw et al. 1979). However, electron microscopy of whole phage and purified phage tail fibers shows a consistently straight rod in this region, suggesting a rigid joining, with no or very little flexibility.
The amino acid sequence of the amino end of the gene 37 protein is highly conserved (as is the corresponding carboxyl end of the gene 36 protein). Figure 3 shows the results of a BLAST-P search using the 60-aa end of the gene 37 protein. This sequence is aligned with the first 37 sequences from the named bacteriophages [matches to “hypothetical phage protein” and bacterial genome sequences (presumably prophage sequences) were excluded]. Of those 60 amino acids, 40 (67%) were invariant and many of the others had conserved changes (e.g., leucine and isoleucine at amino acid I38 in the T4 sequence). No position had more than four different amino acids. Overall, this suggests an interaction structure that has very specific interactions between the gene 36 and 37 proteins to form this joint. It is tempting to speculate that this lack of variability suggests some type of rigid intertwining of both proteins into a rigid joint, but there is, to date, no structural data to support this.

Alignment of the first 60 amino acids of gp37 with other bacteriophage equivalent proteins as identified by BLAST-P. Asterisks indicate positions that are identical in all sequences, black highlighting indicates the amino acid that is present in ≥50% of the sequences, gray highlighting indicates conserved substitutions in >50% of the sequences (Henikoff and Henikoff 1992; Riek et al. 2007)
Beyond these 60 amino acids, there is little sequence homology. As discussed above, the central rod region is highly variable in terms of sequence. There appears to be little interaction between the binding domain end and the central rod region, a notion supported by the isolation of two partial spontaneous deletion mutants of the central rod region. The first one identified, 37SΔ1, has a 346-aa deletion, with the deletion beginning at amino acid D73, just 13 amino acids past the end of the conserved region (Fig. 1c). The tail fibers with the 37SΔ1 deletion appear to function normally (Hyman et al. 2002). A second deletion mutant, 37SΔ2, with a 369-aa deletion beginning at A175, has also been isolated. Although it is not as well characterized, the 37SΔ2 phages appear to grow normally as well. The lack of obvious phenotype by these deletions, each approximately one-third of the complete 1026-aa gene 37 protein, suggest that the upstream and downstream parts of the protein (the 36 binding domain upstream and downstream, the collar, assembly initiation domain, and receptor binding domain) function independently.
The junction-binding region of the distal rod, gp36
The top part of the distal rod is formed by a homo-trimer of gp36, a relatively small protein of 221 residues per monomer. Bio-informatic analysis suggests it does not contain coiled-coil regions and has predominantly a β-structure. Analysis using HHpred (Alva et al. 2016) only predicts similarity for residues 35–61 of gp36 with residues 1234–1250 of gp34. In gp34, these residues are located at the junction of the P5 domain and the C-terminal β-helical region, consisting of a long loop and a short α-helix (Fig. 4; Granell et al. 2017). The α-helices of the three monomers face each other sideways in the gp34 structure, forming a ring with a hydrophobic interior. This similarity, albeit in a short sequence stretch, between the C-terminal part of gp34 and the N-terminal part of gp36, suggests that gp35 may bind both parts in a similar way. Apart from this, no sequence similarity to other trimeric β-structured fiber proteins is apparent. Therefore, the rest of the protein may well contain a new fold.

Structure of the proximal rod of the phage T4 LTF. Side view of a ribbon representation of the gp34 fragment containing part of the P2 and the whole P3, P4, and P5 domains. The three chains of the protein homo-trimer are in green, red, and blue. Residues mentioned in the text are labeled
The junction, gp35
The knee-cap protein, gp35, is a 372-aa protein for which no structural information is available. In contrast to the other three structural proteins of the LTF, it is present as a monomer, and it may well be the protein that induces the fixed-angle bend between the proximal and distal rod regions. An analysis using HHpred suggests the notion that the protein may consist of two carbohydrate-binding domains. Residues 19–178 show homology to PDB entry 5GGN, the N-terminal domain of human protein O-mannose β-1,2-N-acetylglucosaminyltransferase (Kuwabara et al. 2016), and residues 228–344 show homology to PDB entry 4XUP, the N-terminal CBM22-1–CBM22-2 tandem domain from the Paenibacillus barcinonensis XYN10CM protein (Sainz-Polo et al. 2015). The two carbohydrate-binding domains may have evolved to bind the gp34 and gp36 proteins instead of carbohydrates, although it is not known which domain of gp35 binds which protein. The short stretch of putative structural similarity between the N-terminal residues of gp36 and the C-terminal residues of gp34 suggest that these are possible gp35 binding sites. In each case, the gp35 domains may envelop the end of more than one of the gp34 and gp36 monomers, respectively, preventing the binding of more copies of gp35.
The phage-proximal rod: gp34
The proximal rod of the LTFs consists of a homo-trimer of the gp34 protein, which contains 1289 amino acids per monomer. The N-terminal P1 domain of the homo-trimer has a barrel shape and is predicted to contain around 400 residues from each of the three monomers. It is this domain that binds to the gp9 trimer on the outer ring of the baseplate (Taylor et al. 2016), although the molecular details of this interaction are currently unknown. HHpred analysis suggests that this domain may have structural homology to the N-terminal part of another T4 baseplate protein, gp10. Residues 96–291 of gp34 are aligned with residues 49–289 of gp10. In gp10, these residues form two β-sandwich domains, the first of which associates laterally with two equivalent β-sandwiches from two other monomers to form a trimeric structure. The second β-sandwich domain also forms a trimeric structure with two equivalent domains, but longitudinally. In the baseplate, there is a bend between these domains of gp10, while in gp34 they are more likely to be stacked straight on top of each other, probably without flexibility between them.
Unfolding experiments suggest that the C-terminal part of gp34 is important for folding (Granell et al. 2014), which is also true for other viral fiber proteins (van Raaij and Mitraki 2004). High-resolution crystal structures of C-terminal parts of gp34 revealed that residues 744–876 contain three of the repeats that are also present in gp12 (Cerritelli et al. 1996; van Raaij et al. 2001; Taylor et al. 2016; Fig. 4). Each of the repeats contains three central α-helices preceded by intertwined loops. Seven more of these repeats are likely present, starting at residue 438 (Granell et al. 2017). Together, the ten repeats make up the P2 domain of gp34. C-terminal to the repeat region (closer to the knee-cap), amino acids 877–900 form a triple-helical region followed by two turns.
The C-terminal part of the structure of gp34 trimers (residues 901–1270) is a long triple β-helix that is interspersed with three tower domains, i.e., P3, P4, and P5 (Granell et al. 2017). The cores of the tower domains are three anti-parallel β-sheets, one contributed by each of the three monomers, and these sheets are also present in other bacteriophage receptor-binding proteins (Koc et al. 2016). The P4 tower domain is decorated with short loops, while the loops of the P3 tower domain are somewhat longer. The P5 domain is extensively decorated, including several short α-helices and a long β-hairpin that extends towards the C-terminus.
The short tail fiber: gp12
Although the short tail fibers are not part of the LTF, it is of interest to briefly discuss their similarities with the LTF proteins. The short tail fibers are parallel homo-trimers of the gp12 protein, which contains 527 residues per monomer. The N-termini attach to gp10 trimers in the baseplate, while gp11 trimers hold the short tail fibers at a hinge region located about half-way (Leiman et al. 2010; Taylor et al. 2016). The short tail fibers contain a β-structured N-terminal domain similar to the N-terminus of gp11 (Leiman et al. 2000), followed by six of the same repeats present in the rod region of gp34. Residues 290–327 form a triple β-helix (van Raaij et al. 2001), followed by a collar domain and the receptor-binding domain, which contains a single central metal ion as described above (Thomassen et al. 2003). The presence of collar domains in gp10, gp11, gp12, and gp37 suggests that these proteins are evolutionarily related and probably the result of gene duplications.
Conclusions and open questions
The partial structures of the gp37 and gp34 complexes have revealed several new trimeric β-structured folds that may also be present in other viral fiber proteins. Sequence analyses, functional studies, and mutational studies have elucidated the role of several domains of the LTF proteins. The N-terminal part of gp34 is responsible for attachment to the T4 baseplate protein gp9, while the C-terminus binds to gp35. The middle part of gp34 probably only fulfills a structural role, forming a stable rod of optimal length for phage function. Gp35 connects to the C-terminus of gp34 and to the N-terminus of gp36, and probably imposes the 160° angles at which the proximal and distal rods are attached to each other. It may do this with two separate domains, each interacting with one of the protein trimers. Gp34 may also connect to gp36 directly, but we cannot be sure about this. Gp36 forms the upper part of the distal rod, binding to gp37 with its C-terminus. The central region of gp37 probably has a more structural role, like that of the central region of gp34. The “business end” of gp37 is the C-terminal region, which forms a collar domain (D10) and a needle region, at the end of which a small intertwined receptor-binding domain is located.
Remaining questions are the atomic structures of domains currently not resolved at high resolution. Future structural studies by X-ray crystallography and electron microscopy surely will lead to some more surprising folds. How gp34–gp35, gp35–gp36, and gp36–gp37 are joined together is also unknown. Judicious design of expression clones and co-crystallization of the different protein domains that interact with each other should shed light on these questions. High-resolution structures of the individual proteins plus cryo-electron microscopy of intact LTFs may also serve to resolve them. The exact mechanism of how gp57 chaperones the folding of the gp34 and gp37 trimers is unknown, as is the precise way that the gp38 chaperone promotes gp37 assembly. More structural and functional studies of gp57 and gp38 will be necessary to resolve this. The exact location of the receptor-binding site on the gp37 tip and the exact nature of the receptor is currently also unknown, but mutational studies suggest that bacterial LPS binds to the end of the tip domain and that the OmpC protein binds to the side (Washizaki et al. 2016). A high-resolution structure of gp37 bound to the receptor will help to settle this question. Finally, how do the LTFs activate the baseplate? One possibility is that in the free phage, the LTFs cannot bend downwards very much due to steric hindrance. However, when attached to the cell by at least three LTFs (Ares et al. 2014), the Brownian motion occasionally forces the phage away from the bacterium, and the proximal part of the stiff LTFs may be forced into a less favorable, more downward angle, leading to the gp9–gp34 complexes pushing against a switch in the baseplate. This switch involves the baseplate proteins gp10 and gp11, as depicted in Fig. 16 of Leiman et al. (2010). One of the steps involves gp11 releasing its hold on the short tail fiber hinge region. The baseplate may then be forced to change conformation, which triggers the sequential changes leading to outer tail tube contraction (Taylor et al. 2016) and, finally, DNA ejection into the bacterium. The solutions to these questions await future structural studies of the LTF and their detailed interactions with the baseplate.
References
Ackermann HW (2003) Bacteriophage observations and evolution. Res Microbiol 154:245–251
Alva V, Nam SZ, Soding J, Lupas AN (2016) The MPI bioinformatics toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Res 44:W410–W415
Ares P, Garcia-Doval C, Llauro A, Gomez-Herrero J, van Raaij MJ, de Pablo PJ (2014) Interplay between the mechanics of bacteriophage fibers and the strength of virus-host links. Phys Rev E Stat Nonlinear Soft Matter Phys 89:052710
Bartual SG, Garcia-Doval C, Alonso J, Schoehn G, van Raaij MJ (2010a) Two-chaperone assisted soluble expression and purification of the bacteriophage T4 long tail fibre protein gp37. Protein Expr Purif 70:116–121
Bartual SG, Otero JM, Garcia-Doval C, Llamas-Saiz AL, Kahn R, Fox GC, van Raaij MJ (2010b) Structure of the bacteriophage T4 long tail fiber receptor-binding tip. Proc Natl Acad Sci USA 107:20287–20292
Bishop RJ, Conley MP, Wood WB (1974) Assembly and attachment of bacteriophage T4 tail fibers. J Supramol Struct 2:196–201
Bishop RJ, Wood WB (1976) Genetic analysis of T4 tail fiber assembly. I. A gene 37 mutation that allows bypass of gene 38 function. Virology 72:244–254
Branden C, Tooze J (1999) Introduction to protein structure. Garland Publishing, New York
Cerritelli ME, Wall JS, Simon MN, Conway JF, Steven AC (1996) Stoichiometry and domainal organization of the long tail-fiber of bacteriophage T4: a hinged viral adhesin. J Mol Biol 260:767–780
Crawford JT, Goldberg EB (1980) The function of tail fibers in triggering baseplate expansion of bacteriophage T4. J Mol Biol 139:679–690
Earnshaw WC, Goldberg EB, Crowther RA (1979) The distal half of the tail fibre of bacteriophage T4. Rigidly linked domains and cross-beta structure. J Mol Biol 132:101–113
Goldberg E, Grinius L, Letellier L (1994) Recognition, attachment, and injection. In: Karam JD, Kutter E, Carlson K, Guttman B (eds) The molecular biology of bacteriophage T4. ASM Press, Washington, DC, pp 347–356
Granell M, Namura M, Alvira S, Garcia-Doval C, Singh AK, Gutsche I, van Raaij MJ, Kanamaru S (2014) Crystallization of the carboxy-terminal region of the bacteriophage T4 proximal long tail fibre protein gp34. Acta Crystallogr F Struct Biol Commun 70:970–975
Granell M, Namura M, Alvira S, Kanamaru S, van Raaij MJ (2017) Crystal structure of the carboxy-terminal region of the bacteriophage T4 proximal long tail fiber protein Gp34. Viruses 9. https://doi.org/10.3390/v9070168
Hashemolhosseini S, Montag D, Kramer L, Henning U (1994) Determinants of receptor specificity of coliphages of the T4 family. A chaperone alters the host range. J Mol Biol 241:524–533
Henikoff S, Henikoff JG (1992) Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci USA 89:10915–10919
Hu B, Margolin W, Molineux IJ, Liu J (2015) Structural remodeling of bacteriophage T4 and host membranes during infection initiation. Proc Natl Acad Sci USA 112:E4919–E4928
Hyman P, Harrah T (2014) Bacteriophage T4 tail fibers as a basis for structured assemblies. American Society of Mechanical Engineers, New York
Hyman P, Valluzzi R, Goldberg E (2002) Design of protein struts for self-assembling nanoconstructs. Proc Natl Acad Sci USA 99:8488–8493
King J, Laemmli UK (1971) Polypeptides of the tail fibres of bacteriophage T4. J Mol Biol 62:465–477
Koc C, Xia G, Kuhner P, Spinelli S, Roussel A, Cambillau C, Stehle T (2016) Structure of the host-recognition device of Staphylococcus aureus phage varphi11. Sci Rep 6:27581
Kreuzer K, Morrical SW (1994) Initiation of DNA replication. In: Karam JD, Drake JW, Kreuzer KN (eds) Molecular biology of bacteriophage T4. ASM Press, Washington, DC, pp 28–42
Kuwabara N, Manya H, Yamada T, Tateno H, Kanagawa M, Kobayashi K, Akasaka-Manya K, Hirose Y, Mizuno M, Ikeguchi M, Toda T, Hirabayashi J, Senda T, Endo T, Kato R (2016) Carbohydrate-binding domain of the POMGnT1 stem region modulates O-mannosylation sites of alpha-dystroglycan. Proc Natl Acad Sci USA 113:9280–9285
Leiman PG, Arisaka F, van Raaij MJ, Kostyuchenko VA, Aksyuk AA, Kanamaru S, Rossmann MG (2010) Morphogenesis of the T4 tail and tail fibers. Virol J 7:355
Leiman PG, Chipman PR, Kostyuchenko VA, Mesyanzhinov VV, Rossmann MG (2004) Three-dimensional rearrangement of proteins in the tail of bacteriophage T4 on infection of its host. Cell 118:419–429
Leiman PG, Kostyuchenko VA, Shneider MM, Kurochkina LP, Mesyanzhinov VV, Rossmann MG (2000) Structure of bacteriophage T4 gene product 11, the interface between the baseplate and short tail fibers. J Mol Biol 301:975–985
Leiman PG, Shneider MM, Mesyanzhinov VV, Rossmann MG (2006) Evolution of bacteriophage tails: structure of T4 gene product 10. J Mol Biol 358:912–921
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612
Prasad BV, Schmid MF (2012) Principles of virus structural organization. Adv Exp Med Biol 726:17–47
Qu Y, Hyman P, Harrah T, Goldberg E (2004) In vivo bypass of chaperone by extended coiled-coil motif in T4 tail fiber. J Bacteriol 186:8363–8369
Riede I, Drexier K, Eschbach ML (1985) The nucleotide sequences of the tail fiber gene 36 of bacteriophage T2 and of genes 36 of the T-even type Escherichia coli phages K3 and Ox2. Nucleic Acids Res 13:605–616
Riek R, Choe S, Greenwald J, Roosild T, Vega M (2007) Compositions and methods for producing recombinant proteins. The Salk Institute for Biological Studies, La Jolla, CA. Patent application EP20050855418. WITO International Application No. PCT/US2005/046853. Date of application (USA) 21 December 2005
Sainz-Polo MA, Gonzalez B, Menendez M, Pastor FI, Sanz-Aparicio J (2015) Exploring multimodularity in plant cell wall deconstruction: structural and functional analysis of Xyn10C containing the Cbm22-1-Cbm22-2 tandem. J Biol Chem 290:17116–17130
Simon LD, Anderson TF (1967) The infection of Escherichia Coli by T2 and T4 bacteriophages as seen in the electron microscope. I. Attachment and penetration. Virology 32:279–297
Snyder M, Wood WB (1989) Genetic definition of two functional elements in a bacteriophage T4 host-range “cassette”. Genetics 122:471–479
Taylor NM, Prokhorov NS, Guerrero-Ferreira RC, Shneider MM, Browning C, Goldie KN, Stahlberg H, Leiman PG (2016) Structure of the T4 baseplate and its function in triggering sheath contraction. Nature 533:346–352
Thomassen E, Gielen G, Schutz M, Schoehn G, Abrahams JP, Miller S, van Raaij MJ (2003) The structure of the receptor-binding domain of the bacteriophage T4 short tail fibre reveals a knitted trimeric metal-binding fold. J Mol Biol 331:361–373
van Raaij MJ, Mitraki A (2004) Beta-structured viral fibres: assembly, structure and implications for materials design. Curr Opinion Solid State Mater Sci 8:151–156
van Raaij MJ, Schoehn G, Burda MR, Miller S (2001) Crystal structure of a heat and protease-stable part of the bacteriophage T4 short tail fibre. J Mol Biol 314:1137–1146
Ward S, Luftig RB, Wilson JH, Eddleman H, Lyle H, Wood WB (1970) Assembly of bacteriophage T4 tail fibers. II. Isolation and characterization of tail fiber precursors. J Mol Biol 54:15–31
Washizaki A, Yonesaki T, Otsuka Y (2016) Characterization of the interactions between Escherichia coli receptors, LPS and OmpC, and bacteriophage T4 long tail fibers. Microbiology 5:1003–1015
Wood WB, Bishop RJ (1973) Bacteriophage T4 tail fibers: structure and assembly of a viral organelle. In: Fox CF, Robinson WS (eds) Virus research. Academic Press, New York, pp 303–304
Wood WB, Crowther RA (1983) Long tail fibers: genes, proteins, assembly, and structure. In: Mathews CK, Kutter EM, Mosig G, Berget PB (eds) Bacteriophage T4. American Society for Microbiology, Washington, DC, pp 259–269
Acknowledgements
The authors wish to thank the reviewers for their comments which helped improved the paper. We also want to thank Fred Eiserling for the drawing of bacteriophage T4 that appears in the upper part of Fig. 1a.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Paul Hyman declares that he has no conflicts of interest. Mark van Raaij declares that he has no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
This article is part of a Special Issue on ‘Biomolecules to Bio-nanomachines—Fumio Arisaka 70th Birthday’ edited by Damien Hall, Junichi Takagi and Haruki Nakamura.
Rights and permissions
About this article

Cite this article
Hyman, P., van Raaij, M. Bacteriophage T4 long tail fiber domains. Biophys Rev 10, 463–471 (2018). https://doi.org/10.1007/s12551-017-0348-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12551-017-0348-5