
Abstract
Bone collagen is found throughout most of the archaeological record. Under experimental conditions, collagen is apparently preserved as an intact molecule, with amino acid compositions and isotopic profiles only changing when almost all of the protein is lost. The ubiquity of collagen in archaeological bone has lead to the development of the use of collagen peptide mass fingerprints for the identification of bone fragments—Zooarchaeology by Mass Spectrometry (ZooMS). We report a novel, but a simple method for the partial extraction of collagen for ZooMS that uses ammonium bicarbonate buffer but avoids demineralisation. We compared conventional acid demineralisation with ammonium bicarbonate buffer extraction to test ZooMS in a range of modern and archaeological bone samples. The sensitivity of the current generation of mass spectrometers is high enough for the non-destructive buffer method to extract sufficient collagen for ZooMS. We envisage that a particular advantage of this method is that it leaves worked bone artefacts effectively undamaged post-treatment, suitable for subsequent analysis or museum storage or display. Furthermore, it may have potential as a screening tool to aid curators in the selection of material for more advanced molecular analysis—such as DNA sequencing.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Collagen survival
Collagen, the most abundant protein in bone and in dentin, is found widespread in the archaeological record (Higham et al. 2006; Tuross et al. 1980) and can persist in fossils up to at least 600 ka (Buckley et al. 2011), perhaps considerably longer (Schweitzer et al. 2009). Type I (bone) collagen is a triple helix composed of two alpha 1 (COL1A1) and one alpha 2 (COL1A2) chain. COL1A1 is more highly conserved between genera than COL1A2 (Buckley et al. 2009; Buckley et al. 2010). A third alpha chain, COL1A3, has been reported exclusively in fish (Piez 1965).
Unlike DNA, which is typically fragmented to below 70 bp when recovered from bone (e.g. Krause et al. 2010), collagen is sometimes treated as if it remains an intact protein. Radiocarbon and stable isotope preparation methods typically isolate an acid insoluble fraction, gelatinise that fraction by heating in weak acid and subsequently retain a high molecular weight >30 kDa fraction. Covington et al (2008) argue that collagen is stabilised by physical compression of the collagen fibril by mineral or chrome tanning agents, in a manner similar to the role of dehydration considered by Miles and Ghelashvili (1999). These findings are believed to be responsible for the observations that during artificial degradation, there is no perceptible reduction in collagen quality until there is less than 1% of the levels of collagen found in modern bone (Dobberstein et al. 2009). However, archaeological bone finds can contain more than 1% of its original collagen, yet show a signal of degraded collagen (Harbeck and Grupe 2009) according to commonly used diagenetic indicators (van Klinken 1999). Collagen degradation, though more elucidated in the past decades, can still not be fully or straightforwardly explained. Of the biomolecules found in archaeological specimen of direct biological significance, collagen has one of the highest chances of surviving over substantial periods of ancient history and in a quality that is useful for various types of analyses, such as proteomics.
Degraded collagen that has been as significantly altered to be detrimental to stable isotope analysis (Harbeck and Grupe 2009) is less of an obstacle for Zooarchaeology by Mass Spectrometry (ZooMS). In contrast to other methods such as stable isotope analysis, protein mass spectrometry does not rely solely on bulk collagen and is less influenced by collagen quality in data interpretation.
Zooarchaeology by Mass Spectrometry
Ancient collagen integrity means that peptide mass can be used to identify the taxonomic source of archaeological bone samples, an approach termed ZooMS (Buckley et al. 2009). ZooMS is a version of peptide (sometimes protein) mass fingerprinting (Pappin et al. 1993; Henzel and Watanabe 2003): an approach broadly analogous to DNA fingerprinting (Fig. 1). A proteolytic enzyme (most commonly trypsin) is used to cleave proteins at specific motifs or residues, generating peptide fragments of varying lengths, similar to a restriction enzyme on DNA. Differences in primary sequence will result in a variation in mass fragment patterns, and these patterns are used as a fingerprint diagnostic for the original protein. In ZooMS, archaeological collagen peptide masses are matched to either in-house standards or masses predicted from collagen sequences (Buckley et al. 2009). Due to the slow rate of collagen evolution, the taxonomic resolution of ZooMS is typically no better than animal genus, much less accurate than DNA-based methods (c.f. Boyko et al. 2009). However, ZooMS does not require template amplification and peptides are measured ‘as is’, which reduces contamination risks.

Schematic illustration of DNA fingerprinting versus protein mass fingerprinting (PMF). Both methods are based on the principle that the primary sequence holds motifs for restriction enzymes or proteases respectively and that the resulting fragments after digestion differ in size and mass. A point mutation in a DNA sequence, when not interfering with the enzyme motif, will have no effect on the fragment length and therefore not be detected. In the case of PMF, a non-synonymous point mutation will result in a change in the amino acid sequence, which is usually coupled to a mass change (excepting the isobaric residues Leu/Ile), even when the protease cleavage site (C-terminal to arginine and lysine residues in the case of trypsin) is unaltered
Minimally destructive analysis
The original ZooMS method (Buckley et al. 2009; Buckley et al. 2010) uses acid to demineralise bone prior to collagen extraction and mass spectrometric analysis. Despite small sample sizes (typically 1–5 mg of a bone), the method is still destructive as samples are dissolved after acid digestion and gelatinisation. In the case of analysis of worked bone artefacts or a screening tool for subsequent analyses (e.g. DNA, stable isotope or radiocarbon analysis), a milder extraction process would be advantageous.
The current generation of mass spectrometers is capable of detecting femtomoles or attomoles of peptides (Vorm et al. 1994). We therefore speculated that we could leverage this instrumental sensitivity to analyse the small fraction of soluble protein that is reported to persist in ancient bone (Tuross et al. 1988; Collins et al. 2009) without the need for acid digestion. Here, we report our findings in using a warm (65°C) ammonium bicarbonate buffer (pH 8.0) to leach collagen into solution for ZooMS analysis (Fig. 2), enabling worked bone artefacts to be analysed without destructive sampling.

Schematic overview of the warm–water extraction method. 1 A small particle of bone is collected. 2 The sample is incubated for an hour at 65°C in ammonium bicarbonate buffer. 3 After incubation, the supernatant—containing protein and interfering particles—is discarded. 4 The sample is incubated for another hour at 65°C in ammonium bicarbonate buffer. 5 The second extract contains collagen. 6 Trypsin is added to cleave the protein into peptides. 7 The dependant of the primary sequence of collagen, peptides with varied masses, is obtained. 8 These variations are visualised in the mass spectra and a species identity can be assigned, using ZooMS
Method and materials
Materials
All modern samples were from bovid bone meal autoclaved at 133°C for 20 min. Archaeological bone was powdered bovid bone from a Mesolithic–Neolithic site in Rosenhof (Germany) dated to 4900/4800 cal BC and whole bone fragments of bovid bone from mid-ninth to tenth century Coppergate, York (UK); both well preserved (resp. Scheu et al. 2008; Ottoni et al. 2009). For macroscopic comparison and Fourier transform infrared spectroscopy (FT-IR) investigation, fragments of bovid bone from Coppergate (UK) were used. Scanning electron microscopy was performed on early medieval comb tines made from worked antler or bone (not established prior to investigation). See also Table 1.
Ammonium bicarbonate (NH4HCO3) buffer protocol
Samples of bone (<1 mg for modern bone; approximately 5 mg for archaeological bone) were incubated for 1 h at 65°C in 50 mM ammonium bicarbonate [pH 8.0] in a polypropylene eppendorf tube or microplate. In pilot experiments, a range of temperatures from 65°C to 95°C was tested; higher temperatures yielded poorer signals in mass spectrometric analysis with modern bone (results not shown). Samples were briefly centrifuged and the supernatant discarded. The extraction was repeated, and this supernatant was collected for incubation with 1 μg/μl sequencing grade modified porcine trypsin (Promega, Southampton, UK) at 37°C for18 h. See also Fig. 2.
Cold acid protocol
Samples (<1 mg for modern bone; approximately 5 mg for archaeological bone) were incubated in 0.6 M HCl for 24 h at 4°C. Demineralised samples were centrifuged, and the supernatant discarded. The acid insoluble fraction was washed in ultrapure water to neutral pH, then incubated for 3 h in 50 mM ammonium bicarbonate [pH 8.0] at 70°C. The supernatant was collected and incubated for 18 h with 1 μg/μl sequencing grade modified porcine trypsin (Promega) at 37°C.
Peptide purification by solid phase extraction
The tryptic digest was filtered over C18 resin (Millipore, Durham, UK; Porvair, Leatherhead, UK) to desalt and concentrate peptides by washing with 0.1% trifluoric acid (TFA). Peptides were eluted in a final volume of 10 μl of 50% acetonitrile (ACN)/0.1% TFA (v/v). One microlitre of elute was mixed on a ground steel plate with 1 μl α-cyano-4-hydroxycinnamic acid matrix solution (1% in 50% ACN/0.1% TFA (v/v/v)) and dried to air.
Mass spectrometric analysis
Each sample was analysed in reflector mode using a calibrated Ultraflex III (Bruker Daltonics, Bremen, DE) MALDI-TOF instrument to measure mass to charge ratios (m/z) of trypsinated fragments. Spectra were analysed using flexAnalysis software v. 3.0 (Bruker Daltonics). The indexed peptides were identified manually according to Buckley et al (2009).
Extraction efficiency measured by Bradford assay
Untrypsinated material (M, H, P—abbreviations in Table 1) extracted by either cold acid or NH4HCO3 buffer method was collected and diluted 10 and 20 times in 50 mM of ammonium bicarbonate [pH 8.0]. One volume of Coomassie Plus Protein Assay Reagent (Thermo Fischer Scientific, Loughborough, UK) was added to each sample and allowed to incubate 1 min at room temperature. Samples were calibrated against standard range of 50–0.8 mg/ml BSA, and a 50 mM ammonium bicarbonate blank. All samples were measured in triplicate using a FLUOStar OPTIMA microplate reader (BMG Labtech, Offenburg, DE) in absorbance mode, with a BMG 0308A filter at a wavelength range of 570–585 nm.
Macroscopic comparison
Two bone particles of approximately equal weight (approximately 70 mg) and size (10 mm diagonal) from the same archaeological bovine sample (H) were treated with either the cold acid method or the NH4HCO3 buffer method. After each incubation step, the samples were weighed, measured and photographed. Concluding regular extraction, the samples were further exposed to three cycles of 12 h at −20°C and 12 h at room temperature in 50 mM ammonium bicarbonate buffer after normal sample treatment to examine the impact of freeze-thaw damage (i.e. repeated removal from a freezer).
Scanning electron microscopy
Comb tines from a worked bone artefact (W) were treated with the NH4HCO3 buffer method. One of these was exposed to a freeze-thaw cycle, due to storage conditions. A third comb tine was left untreated as a control.
All three samples were examined by scanning electron microscopy. Samples were suspended in a copper shim and viewed uncoated at 5 kV on a JSM 6490LV microscope (JEOL, Tokyo, Japan).
For (worked) bone surfaces ultrasonic cleaning in the presence of a mild detergent is recommended (Rose 1983) to remove dirt from the surfaces. However, the NH4HCO3 buffer method involves a similar washing step, obviating the need for potentially destructive cavitation effects of ultrasound. Samples were cleaned with cotton wool, but following extraction with NH4HCO3 macroscopic surface impurities were still visible.
Fourier transform infrared spectroscopy
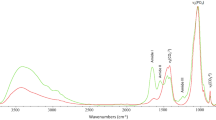
Bone apatite powder (2 mg) was ground with 200 mg of spectroscopic grade KBr. The powder was placed within a 12-mm disc and pressed into a pellet (3 mm window) using a hydraulic press at 10,000 psi. Pellets were scanned 16 times using a Perkin-Elmer FT-IR spectrometer. Spectra were recorded from 4,000 to 400 cm−1 and baseline corrected.
Peak heights at wave numbers 565 (v 4 PO4) and 605 (v 4 PO4) and the valley at ∼590 cm−1 between peaks 565 and 605 cm−1 were measured to calculate the infrared splitting factor (SF). The peak heights at wave numbers 1035 (v 3 PO4) and 1,415 cm−1 (v 3 CO3) were measured for calculation of carbonate to phosphate ratio (CO3/PO4). These are two common semi-quantitative measurements used to evaluate diagenesis in apatite. Additionally, the collagen content can be estimated by calculating the ratio of intensities of the amide peak at 1,640 cm−1 and the phosphate peak at 1,035 cm−1 (Trueman et al. 2008).
The SF evaluates the crystalline structure of bioapatite. For modern bone, SF ranges between 2.5 and 2.9 (Wright and Schwarcz 1996). An increase in SF indicates re-crystallisation and an increase in crystal size and order. This is generally coupled with a decrease in CO3/PO4, i.e. a loss in carbonate (Sillen 1989). Measurements made on fresh cattle bone gave an amide to phosphate ratio (Am/PO4) of 0.36. Any values below this would indicate a relative decrease in collagen content.
Results
Comparison on modern bone
Autoclaved, modern animal bone extracted using both the acid and NH4HCO3 buffer method produced sufficient collagen for MALDI-MS analysis and identification of indexed peptides for speciation (Fig. 3a). The buffer method obtains reliable results from bone meal, presumably extracting a fraction of collagen solubilised by the heat and pressure of the autoclave. Similar results are obtained from archaeological material (H in Table 1), showing that both methods can be used for ZooMS applications (Fig. 3b).

MALDI-TOF spectra from bovid (Bos taurus) bone extracts. a Modern bone (M, see Table 1), demineralised (above) and non-demineralised (below). b Archaeological bone (H, see Table 1), demineralised (above) and non-demineralised (below). Assigned in capital letters, A–G, are indexed peptides as designated by Buckley et al. (2009). t is used to indicate a trypsin autocleavage product
To assess the amount of protein extracted in both protocols, first and second extracts from the bone of three different ages (M, H, P; Table 1) were determined by Bradford assay. The highest yield of protein with the Bradford assay was always obtained in the first extract; concentrations decreased with subsequent extractions. However, MALDI-TOF-MS analysis of the first protein extract has consistently lower peak intensities and ion currents than the following extract. Further repeated extractions still produced sufficient protein for identification, although below the detection limit of the Bradford assay. There was no significant difference in yields of detectable protein with either the acid or ammonium bicarbonate methods. However, in archaeological samples, more protein is obtained using acid demineralisation, in both the first and any subsequent incubation. Estimated yields (Bradford assay) are shown in Table 2 for each sample per milligramme of original bone weight.
Macroscopic comparison on artefact preservation
Figure 4 shows the effect of each incubation step on bone material (H). Untreated, both samples are of comparable size and weight. After acid treatment, sample A was decreased by 76% in weight, indicating that all the mineral (accounting for 63% by weight; Glimcher and Krane 1968), and some of the collagen fraction was lost. A transparent collagen “ghost” was left after drying overnight to air. Ultimately, sample A lost approximately 90% of its original weight (74.3 mg) and decreased 50% in size (11 mm). Sample B showed no significant decrease in either size (12 mm) or weight (59.9 mg). After multiple freeze-thaw cycles, no measurable changes were observed in either sample.

Macroscopic comparison of the cold acid treatment versus the NH4HCO3 buffer method on well-preserved archaeological bovid bone (H, see Table 1). In the original frame, both samples are untreated. After demineralisation, sample A (acid) has been incubated for 24 h in 0.6 M HCl. Sample B (buffer) is still untreated. In the last extraction, sample A has been incubated for 3 h at 70°C in 50 mM NH4HCO3 buffer. Sample B has been incubated twice for 1 h at 65°C in NH4HCO3 buffer. Percentages of size and weight changes have been plotted below
Microscopic and molecular comparison on artefact preservation
Scanning electron microscope images of untreated and treated artefacts (W) with the NH4HCO3 buffer method show no marked differences between bone surfaces (Fig. 5). Both the untreated sample and the treated samples (including one stored in a freezer) show surface damage, which may be caused by manufacture, use or taphonomy; the detail on these does not appear to change following extraction.

SEM images of medieval comb tines (W, see Table 1). Left to right: untreated control, sample treated with NH4HCO3 buffer, sample treated with NH4HCO3 buffer and exposed to a freeze-thaw cycle. Above magnification at ×200; Below magnification at ×1,500
There is no evidence that the NH4HCO3 method measurably alters bone exposed to incubation. Acid extraction, unsurprisingly leads to changes in the mineral phase—specifically a lower IR-SF (i.e. a lower crystallinity), a higher proportion of carbonates and a high amide to phosphate ratio (indicating a relative increase in collagen over mineral).
The SF, CO3/PO4 and Am/PO4 values of the NH4HCO3 buffer-extracted sample are similar to the control values, and both are within the typical range for modern bone. However, the dissolution of apatite has reduced the crystallinity after cold acid treatment (Table 3). Low errors of SF values are also typical for unaltered bone.
Discussion
A recent paper (Dobberstein et al. 2009) argued, on the basis of CNBr digestion, that bone collagen remains largely intact in archaeological bone. Given the high temperature (>150°C) required to denature intact collagen (Kronick and Cooke 1996), the mild extraction procedure (2 × 1 h at 65°C) would be too low to fully denature undamaged collagen, yet yields repeatable and detectable results for ZooMS analysis in both modern and archaeological bone. A small, but significant soluble fraction is present in most bone through accumulated damage (Collins et al. 2009), and despite low yield, the coupling of concentration and purification by solid phase extraction and the sensitivity of MALDI-TOF-MS means that NH4HCO3 buffer extraction is sufficient for ZooMS analysis. We are currently exploring a larger body of samples, including material with varying thermal history, as the ability to release the minimal fraction of soluble collagen may be limited in some burial environments. The soluble fraction may have been leached out completely prior to investigation if the inhumation period was exposed to an extensive water influx.
Even with soluble collagen so easily flushed out, we consider the risk of cross contamination during the inhumation period low. Due to the lack of amplification, there is little risk in a bone-preserving environment in which endogenous collagen would not exceed exogenous collagen by such a measure that contamination would show in mass spectra following this method.
Sample interference
Despite the fact that protein yields were highest in the first extract and decreased with each subsequent extraction (as estimated by Bradford assay and SDS-PAGE gel), the first extract invariably had lower peak intensity in mass spectra than the second extract. Subsequent extractions had progressively lower peak intensity; a pattern observed in both modern and archaeological bone.
It is unclear why the first extract has sub-optimal performance, but we suspect that this extract is enriched with compounds that compete in solid phase extraction with the peptides, co-crystallise with the matrix or otherwise prevent efficient ionisation of peptides. The problem is more apparent in modern than archaeological extracts. The interfering component was probably moderately hydrophobic, given that the interference could be somewhat reduced (but not excluded) if extraction was undertaken in polypropylene microplates rather than polystyrene (Sambrook and Russell 2001). One possible candidate is the phospholipids; their rapid reduction in archaeological samples (Millard 2001; Evershed et al. 1995) would in part explain the lower interference in archaeological materials. However, we suspect that a more complex range of compounds is responsible as interference was also present in all archaeological samples, irrespective of age and burial environment.
Artefact preservation and method potential
Our results show that in the case of well-preserved bone, the NH4HCO3 buffer extraction does not measurably alter the material. This allows worked artefacts to be investigated by ZooMS without destroying the sample and the artefact is only exposed twice to a weak (50 mM) ammonium bicarbonate buffer of pH 8.0 at 65°C for an hour (and this could be used as part of the cleaning process). It is unlikely that these conditions will otherwise modify any object of mineralised tissue, as diagenetic parameters remain unaltered after treatment. However, any major changes in crystallinity in bone before buffer extraction may have an effect similar to cold acid extraction and potentially have a destructive effect.
With the NH4HCO3 buffer extraction, the speed and sample throughput were vastly improved, while keeping overall costs down. Ultimately, the extraction after cold acid treatment, by obtaining a higher protein yield, will have a higher efficiency and consequently a greater success rate. However, the partial leaching in the NH4HCO3 buffer extraction allows the analyst to revisit the same specimen, and at the same time, to investigate a higher number of samples at lower cost, as it obviates the need for a neutralisation step.
Conclusion
We have demonstrated the use of a mild NH4HCO3 buffer-based extraction method for collagen from mineralised tissue. The NH4HCO3 buffer extraction yields sufficient collagen from well-preserved mineralised tissue for mass spectrometric analysis and ZooMS identification of species. Potentially, the method can be employed to safely identify valuable artefacts or tiny fragments. It is also possible to revisit an analysis on the same material and furthermore the sample is not compromised if subsequent destructive analysis is required.
References
Ashby SP (2009) Combs, contact and chronology: reconsidering hair combs in early-historic and Viking-Age Atlantic Scotland. Mediev Archaeol 53:1–33
Boyko AR, Boyko RH, Boyko CM, Parker HG, Castelhano M, Corey L, Degenhardt JD, Auton A, Hedimbi M, Kityo R, Ostrander EA, Schoenebeck J, Todhunter RJ, Jones P, Bustamante CD (2009) Complex population structure in African village dogs and its implications for inferring dog domestication history. PNAS 106(33):13903–13908
Buckley M, Collins MJ, Thomas-Oates J, Wilson JC (2009) Species identification by analysis of bone collagen using matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry. Rapid Commun Mass Spectrom 23:3843–3854
Buckley M, Kansa SW, Howard S, Campbell S, Thomas-Oates J, Collins MJ (2010) Distinguishing between archaeological sheep and goat bones using a single collagen peptide. J Archaeol Sci 37:13–20
Buckley M, Larkin M, Collins MJ (2011) Mammoth and Mastodon collagen sequences: survival and utility. Geochim Cosmochim Acta 75(7):2007–2016
Collins MJ, Penkman K, Rohland N, Shapiro B, Dobberstein RC, Ritz-Timme S, Hofreiter M (2009) Is amino acid racemization a useful tool for screening for ancient DNA in bone? Proc R Soc B 276:2971–2977
Covington AD, Song L, Suparno O, Koon HEC, Collins MJ (2008) Link-lock: an explanation of the chemical stabilisation of collagen, vol 92. Society of Leather Technologists and Chemists, Dewsbury
Dobberstein RC, Collins MJ, Craig OE, Taylor G, Penkman K, Ritz-Timme S (2009) Archaeological collagen: why worry about collagen diagenesis? J Archaeol Anthropol Sci 1:31–42
Evershed RP, Turner-Walker G, Hedges REM, Tuross N, Leyden A (1995) Preliminary results for the analysis of lipids in ancient bone. J Archaeol Sci 22:277–290
Glimcher MJ, Krane SM (1968) The organization and structure of bone and the mechanism of calcification. In: Ramachandran GN, Gould BS (eds) Treatise on collagen: biology of collagen, vol 2b. Academic Press, London, pp 67–251
Harbeck M, Grupe G (2009) Experimental chemical degradation compared to natural diagenetic alteration of collagen: implications for collagen quality indicators for stable isotope analysis. J Archaeol Anthropol Sci 1:43–47
Henzel WJ, Watanabe C (2003) Protein identification: the origins of peptide mass fingerprinting. J Am Soc Mass Spectrom 14:931–942
Higham TFG, Jacobi RM, Bronk Ramsay C (2006) AMS radiocarbon dating of ancient bone using ultrafiltration. Radiocarbon 48(2):179–195
Krause J, Briggs AW, Kircher M, Maricic T, Zwyns N, Derevianko A, Pääbo S (2010) A complete mtDNA genome of an early modern human from Kostenki, Russia. Curr Biol 29(3):231–236
Kronick PL, Cooke P (1996) Thermal stabilization of collagen fibers by calcification. Connect Tissue Res 33(4):275–282
Miles CA, Ghelashvili M (1999) Polymer-in-a-box mechanism for the stabilization of collagen molecules in fibers. Biophys J 76:3243–3252
Millard AR (2001) The deterioration of bone. In: Brothwell DR, Pollard AM (eds) Handbook of archaeological sciences. Wiley, Chichester, pp 637–647
Ottoni C, Koon H, Collins MJ, Penkman K, Rickards O, Craig OE (2009) Preservation of ancient DNA in thermally damaged archaeological bone. Naturwissenschaften 96:267–278
Pappin DJC, Hojrup P, Bleasby AJ (1993) Rapid identification of proteins by peptide-mass fingerprinting. Curr Biol 3:327–332
Piez KA (1965) Characterization of a collagen from codfish skin containing three chromatically different alpha chains. Biochemistry 4:2590–2596
Rose JJ (1983) A replication technique for scanning electron microscopy: applications for anthropologists. Am J Phys Anthropol 62:255–261
Sambrook J, Russell DW (2001) Protocol 1: DNA transfection mediated by lipofection. In: Molecular cloning: a laboratory manual, volume 3, vol 3. Cold Spring Harbor Laboratory Press, New York, p 16.19
Scheu A, Hartz S, Schmoelcke U, Tresset A, Burger J, Bollongino R (2008) Ancient DNA provides no evidence for independent domestication of cattle in Mesolithic Rosenhof, Northern Germany. J Archaeol Sci 35:1257–1264
Schweitzer MH, Zheng W, Organ CL, Avci R, Suo Z, Freimark LM, Lebleu VS, Duncan MB, Vander Heiden MG, Neveu JM, Lane WS, Cottrell JS, Horner JR, Cantley LC, Kalluri R, Asara JM (2009) Biomolecular characterization and protein sequences of the Campanian Hadrosaur B. canadensis. Science 324:626–631
Sillen A (1989) Diagenesis of the inorganic phase of cortical bone. In: Price TD (ed) The chemistry of prehistoric human bone. Cambridge University Press, Cambridge
Trueman CN, Privat K, Field J (2008) Why do crystallinity values fail to predict the extent of diagenetic alteration of bone mineral? Palaeogeography, Palaeoclimatology, Palaeoecology 266(3–4):160–167
Tuross N, Eyre DR, Holtrop ME, Glimcher MJ, Hare PE (1980) Collagen in fossil bones. In: Hare PE, Hoering TC, King K Jr (eds) Biogeochemistry of amino acids. Wiley, New York, pp 53–64
Tuross N, Fogel ML, Hare PE (1988) Variability in the preservation of the isotopic composition of collagen from fossil bone. Geochim Cosmochim Acta 52(4):929–935
van Klinken GJ (1999) Bone collagen quality indicators for palaeodietary and radiocarbon measurements. J Archaeol Sci 26:687–695
Vorm O, Roepstorff P, Mann M (1994) Improved resolution and very high sensitivity in MALDI TOF of matrix surfaces made by fast evaporation. Anal Chem 66:3281–3287
Wright LE, Schwarcz HP (1996) Infrared and isotopic evidence for diagenesis of bone apatite at Dos Pilas, Guatemala: palaeodietary implications. J Archaeol Sci 23(6):933–944
Acknowledgements
We would like to thank Meg Stark for the assistance with acquiring SEM images and Isabella von Holstein for the additional assistance in practical work and Terry O’Connor, Uli Schmölcke, Scott Reaney and Steve Ashby for the donated samples. The project is a part of LeCHE and funded by Marie Curie FP7 Framework (MC-ITN 215362 LeCHE); Matthew Collins is supported by the EU SYNTHESYS II Project (JRA1). The Ultraflex III was used courtesy of the York Centre of Excellence in Mass Spectrometry. The York Centre of Excellence in Mass Spectrometry was created thanks to a major capital investment through Science City York, supported by Yorkshire Forward with funds from the Northern Way Initiative.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
van Doorn, N.L., Hollund, H. & Collins, M.J. A novel and non-destructive approach for ZooMS analysis: ammonium bicarbonate buffer extraction. Archaeol Anthropol Sci 3, 281–289 (2011). https://doi.org/10.1007/s12520-011-0067-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12520-011-0067-y