
Abstract
Background
Kaposiform hemangioendothelioma (KHE) is a rare vascular tumor affecting infants and young children. Although benign, it can be associated with an aggressive locally growing tumor and/or a life-threatening Kasabach–Merritt phenomenon (KMP). To date, only reviews of limited cases have been performed. We, therefore, conducted a comprehensive literature search to collect relevant data and make recommendations for future treatment trials.
Methods
Review of the available literature between 1993 and 2017 revealed a total of 105 publications involving 215 patients of less than 21 years of age. To this, we added 12 from our department and 4 from the Cooperative Weichteilsarkomstudie database.
Results
We found that KMP was present in 79% of the infants, in 47% of the 1–5-year olds, in 43% of the 6–12-year olds, and in 10% of the 13–21-year-old patients. KMP was present in nearly all (94%) patients with retroperitoneal tumors and in all patients with extra-regional tumors. The median size of a KHE without KMP was 12 cm2 as compared to 49 cm2 when associated with a KMP. With complete (not further classifiable if R0 or R1) resection, all patients were cured. If inoperable, response regarding KMP/regression of tumor size was seen in 29/28% with steroid-, 47/39% with vincristine-, 44/43% with interferon alpha-, 65/61% with anti-platelet agents-, and in 97/100% with sirolimus-containing therapies.
Conclusions
Patients with progressive KHE should undergo resection whenever it is considered a safe option. If inoperable, sirolimus should be the first choice for treating KMP and reducing tumor size.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
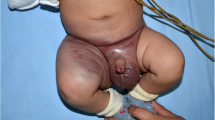
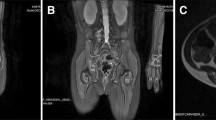
Kaposiform hemangioendothelioma (KHE) is a rare vascular tumor affecting infants and young children that presents as an infiltrative growing, erythematous or purpuric soft tissue mass [1]. Tumors can be located superficially in the skin or deeper in the extremities, head/neck, trunk, retroperitoneum, mediastinum, bone or other organs [1]. Multifocality has also been described [2]. Although benign, these tumors can be associated with severe morbidity and mortality. Due to abnormal growth of capillary endothelial cells, up to 70% of the patients suffer from a Kasabach–Merritt phenomenon (KMP) with thrombocytopenia and coagulopathy [3]. Up to 30% of the affected patients die often as a result of the associated coagulopathy [1, 3, 4]. The association between a vascular tumor and a KMP has thus far only been detected in patients with KHE and those with tufted angioma [5] (now actually recognized as a continuum of the same vascular tumor [4, 6]).
To distinguish KHE from hemangioma or other vascular malformations or tumors, a histopathologic evaluation should be carried out. However, if KMP is present and there is a potential risk for bleeding, then biopsy may be omitted since KMP is exclusively secondary to KHE or tufted angiomas but not to infantile hemangiomas [5]. KHE is characterized by a vascular spindle-shaped cell proliferation and is similar to Kaposi sarcoma-like fascicular cells and their surrounding lymphatic vessels as first described by Zuckerberg et al. [3, 7, 8].
In contrast to hemangioma, KHE only infrequently undergoes spontaneous regression. Even with effective treatment, the tumors never totally regress [9, 10]. As KHE is potentially life threatening and there is the possibility of tumor progression if untreated, early intervention is mandatory. However, since KHE is rather rare and with only case reports and only one smaller metaanalysis published to date, no evidence-based treatment strategies have yet been established. The most important treatment options discussed are steroids, vincristine, interferon alpha and sirolimus [3, 4, 11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26]. Whereas steroids as single agents have been rather ineffective with low response rates up to about 10-30% [11, 27, 28], there are reports that vincristine is much more effective [11, 12, 15] with response rates between 62 and 100% [11, 14, 15, 27, 28]. In 2013, a consensus statement was published, recommending vincristine 0.05 mg/kg/week and steroids (oral prednisolone 2 mg/kg/day or i.v. methylprednislone 1.6 mg/kg/day) for patients with KMP and oral prednisolone for patients with growing tumor or symptoms not due to KMP [4]. An excellent long-term outcome regarding KMP had also been reported as a result of adding aspirin and ticlopidine to vincristine (VAT), although no effect was seen in reduction of tumor size [13]. Recent publications reported sirolimus as safe and highly efficient in treating KMP and achieving tumor shrinkage [21,22,23,24,25, 29,30,31].
To learn more about KHE, both with and without the KMP, we conducted a literature search and combined these data with that from our own patients. Information was collected with regard to localisation, tumor size, the presence of KMP, treatment modality, response and long-term outcome.
Methods
Data were collected retrospectively from 12 children from our own Department for Pediatric Hematology and Oncology in the Dr. von Hauner Children's Hospital and 4 children from other participating centers of the Cooperative Weichteilsarkomstudie (CWS) database in Germany.
To get more information about KHE with and without KHP, we conducted a systematic literature review in the medline database (http://www.ncbi.nlm.nih.gov/pubmed/). We identified a further 215 affected children from a review of 105 papers published between 1993 and December 2017. We started our literature search in 1993 since Zukerberg et al. first characterized KHE as a separate tumor entity at that time [7]. Inclusion criteria were histologically proven KHE, age below 21 years and publications written in German or English language.
We collected the following data: age, sex, localisation and size of the tumor, presence of KMP, therapy, response and outcome. Tumor localisation was defined as follows: extremities, trunk, head/neck, retroperitoneum, mediastinum, organ related (e.g., heart, kidney, gut, pancreas) and bone. Larger than 1 region (> 1 region) means that the tumor exceeds the edges of the original organ (T4 according to the TNM classification).
To evaluate the size of the tumor, the surface area was calculated. When multiple measurements were given, the biggest stated dimension was used. Depth-related information was only provided in a few cases and, thus, this could not be included in the analysis.
KMP was defined as positive when thrombocytopenia (< 100 × 103/µL) and abnormalities in coagulation parameters were reported.
Statistical methods
For statistical analysis, PRISM 6.0 (GraphPad, San Diego, USA) was used. P values were calculated using the non-parametric Mann–Whitney U test and the two-tailed Fisher`s exact test. P values were not calculated where only purely descriptive data was available. Response was defined 1) for KMP: resolvement of the coagulation disorder and 2) for tumors: any shrinkage of the size of the tumor. Evaluation according the RECIST (Response Evaluation Criteria in Solid Tumors) criteria was not possible due to missing or incomplete datasets. For outcome, the following items were defined: ANED: alive with no evidence of disease, ARD: alive with residual disease, AWD: alive with disease, DOD: died of disease, DUC: died of unknown/unrelated cause.
Results
KHE and age-dependent tumor location
The median age at diagnosis, age distribution, sex, and tumor location for the 215 patients from the literature review and the 16 patients from the Munich/CWS study group are shown in Table 1. About 2/3 of all patients were infants (66% of 231 patients) with decreasing numbers with older age. In infants (153 patients), most tumors were located in the extremities (25%), trunk (29%) and head/neck (18%). In toddlers (1–5 years), KHE was mostly located in the head/neck area (47% of 43 patients). With increasing age, most tumors were found in the extremities again. Organ related, retroperitoneal or mediastinal tumors were only found in those less than 6 years old.
KMP and age and location
KMP was found in about 2/3 of all patients (67% of 231) and 68 (29%) patients had no KMP (Table 2). The older the patient the lower is the incidence of KMP. KMP was diagnosed in 79% of 142 infants, in 47% of 38 toddlers (1–5 years), in 43% of 21 patients (6–12 years) and in 10% of 10 adolescents (13–21 years). The median age of the patients with KMP was 2 months and without KMP 22.5 months (P < 0.0001) (data not included).
Nearly, all children with tumor located in the retroperitoneum (15/16), with multifocality (6/7) or region exceeding tumors (9/9) had a KMP. None of the 5 patients with KHE within the bone without other tissue involvement had a KMP. Tumors located within the extremities (65% of 63 patients), trunk (77% of 53 patients), mediastinum (67% of 9 patients) or head/neck (58% of 57 patients) had a similar incidence of a KMP.
KMP and size
The next question was whether the size of the tumor has an impact on the incidence of KMP. Forty-four patients had sufficient information available to calculate tumor size. The median tumor size for these patients was 27 cm2 (range 1–600 cm2). There was a clear correlation between the appearance of KMP and increasing tumor size. KHE without KMP had a median size of 12 cm2 (25%-quantile 4.0 cm2–75%-quantile 26.85 cm2) and KHE with KMP 48.75 cm2 (25%-quantile 24.0 cm2–75%-quantile 82.5 cm2) (P = 0.0003) (Fig. 1).

Kaposiform hemangioendothelioma (KHE) without Kasabach-Merritt phenomenon (KMP) had a median size of 12 cm2, KHE with KMP 49 cm2 (n = 44) (P = 0.0003)
Treatment options
The most interesting question is how to treat these patients. The strategies reported can be divided into 3 groups: 1) resection/interventional procedures, embolization or ligation, 2) irradiation and 3) drugs (especially those with anti-angiogenetic effects, chemotherapy agents or coagulation affecting substances) (Table 3).
The most frequently used strategies were (in descending order): steroids, vincristine, interferon alpha, resection, platelet aggregation inhibitors, sirolimus, embolization and propranolol. In most cases, a combination of different strategies was used. The most important combinations were steroids/vincristine (in 38 cases), steroids/interferon alpha (23 cases) and vincristine/aspirin/ticlopidin (VAT, 16 cases).
Therapeutic response in KHE and KMP
In 13 patients (6% of 231 patients), no treatment was given. Two infants both with retroperitoneal tumors died, one at the age of 8 days due to intracranial bleeding and the other in utero. The remaining 11 patients had a tumor located in the extremities (4 patients), trunk (4 patients), head/neck (2 patients) and organ related (1 patient: intracardial). None of these patients had a KMP. Two patients (one with a head/neck tumor and one with a trunk tumor) had a spontaneous regression of the tumor and the other 9 had a stable disease or slight tumor progression within the recorded observation times of between 6 months to 6 ½ years.
In 18% of 231 children, a complete resection was possible. Fourteen of those 42 had a KMP. With complete resection, 38 patients were reported as cured with no relapse. For the remaining 4 patients, data for response and outcome are missing.
With steroids, 29% (monotherapy 32%, in combination 30%) of the children responded regarding KMP and 28% (monotherapy 26%, in combination 28%) of children showed tumor shrinkage (Table 4). For vincristine, 47% (monotherapy 78%, in combination 43%) responded with respect to the KMP and 39% (monotherapy 40%, in combination 39%) with respect to regression of tumor size.
With interferon alpha, 44% (monotherapy 92%, in combination 27%) achieved a response regarding KMP and 43% (monotherapy 75%, in combination 33%) achieved regression of the tumour size. Embolization, radiotherapy and propranolol had a KMP response and tumor shrinkage in about 1/3 of the small studies performed (13–20 cases each). Aspirin as anti-platelet agent either in combination with ticlopidin or with dipyridamol had a response in about 2/3 of the 23 cases involved.
There were 34 patients treated with sirolimus, 29 with monotherapy, 5 in combination with vincristine or steroids or vincristine combined with cyclophosphamide and steroids. Thirty of the evaluable 31 patients had a response in KMP and all 34 in tumor shrinkage. Of note is that the one patient with no response only had sirolimus for 7 days.
Comparison of the effect of steroids, vincristine, interferon alpha and sirolimus on tumor size
Comparison of monotherapies of interferon alpha, vincristine and sirolimus to steroids (Table 5) demonstrated a highly significant treatment benefit of sirolimus with respect to tumor shrinkage. Interferon alpha was also significantly better than steroids. There was no difference between vincristine and steroids.
Comparison of the effect of steroids, vincristine, interferon alpha and sirolimus on response of KMP
Monotherapy treatment with interferon alpha, vincristine and sirolimus all had a significantly higher probability of KMP response as compared to steroid monotherapy (Table 6).
Long-term outcome
Long-term outcome could be evaluated in 191 of the 231 patients with a median observation time of 18.5 months (range 0 months–16 years). Of the 191 patients, 152 survived with no evidence of disease or with residual non-active tumor (80%) (Table 7). Eighteen (9% of 191) of the children died due to complications of the disease including intracranial bleeding (2), disseminated intravascular coagulation (3), bleeding (2), intra-abdominal bleeding (1), pneumonia due to aspiration (1), progression (1), intrauterine death (1), sepsis (2) and compression of the respiratory tract (1). In 5 cases, the cause of death was not specified. One more patient died of unknown causes but not due to KHE or KMP.
The prognosis was worst for patients with KHE located in the retroperitoneum; 4 of the 14 patients died due to the disease. The outcome was not different between the different age groups (data not shown). 10 vs. 7% died in the group with vs. without KMP.
Discussion
In this retrospective review of 231 children with KHE, our special interest was to collect data to learn more about KHE. As a result of this work, we were able to confirm the infant predominance [1, 11, 12], since about 2/3 of our patients with KHE were infants. The numbers decreased with increasing age.
In infants, about 1/3 of the tumors were located either in the extremities (25%) or in the trunk (29%) and about 20% in the area of the head and neck. In toddlers (1–5 years), about 50% of the tumors were found in the area of the head and neck. Organ-related, retroperitoneal or mediastinal tumors were only found in patients younger than 6 years.
Our review also confirmed that infants and toddlers have a higher risk of developing a KMP as compared to older children [1]. We found that KMP was present in 79% of infants, 47% of 1–5-year olds, 43% of 6–12-year olds, and 10% of 13–21-year olds.
There are special tumor presentations, which are associated with a higher incidence of KMP. Nearly all children with tumors located in the retroperitoneum or with multifocality and all patients with region exceeding tumors had a KMP. Sarkar et al. [5] found that retroperitoneal tumors were associated with a high mortality up to 24% because of bleeding or tumor invasion. Croteau et al. [1] showed that retroperitoneal and intrathoracic KHE had a KMP in 85 and 100% of cases, respectively. No incidences of KHEs limited to the bone (no other organ involvement) and KMP were found in this analysis. This was also reported by Croteau et al. [1]. In our analysis, tumors located in the extremities, trunk or head/neck had a similar incidence of KMP (65 vs 77 vs 58%).
There is a significant correlation between the size of the tumor and the incidence of KMP. We found that a KHE without KMP had a median size of 12 cm2, whereas a KHE with KMP had a median size of 49 cm2. Gruman et al. [32] demonstrated the association of increasing tumor size and incidence of KMP. However, this finding does not allow any predictions, since there were patients in our database with big tumors without KMP and vice versa. In addition, a KHE can grow rapidly with a sudden appearance of a KMP.
Treatment of all patients with growing or symptomatic tumors and/or KMP should be started as soon as possible. We found that 9% of all patients died due to the disease. This is was often due to the coagulation disorder since 8/18 died as a result of bleeding. However, we could not find a difference in mortality between patients with KMP (10%) and those without (7%).
From our data, we may conclude that all patients (including those who are asymptomatic) with multifocality, with retroperitoneal tumors, with tumors exceeding one region and who are less than 6 years old should receive treatment independent of tumor size as these patients have a very high risk of developing a KMP. All other patients (i.e., > 6 years old, asymptomatic tumors outside of the peritoneum or retroperitoneal space, tumors confined to one region) may benefit from a watch-and-wait strategy with strict control intervals.
The best treatment option is complete resection as this was curative in all patients studied. Thus, whenever possible, all patients with progressive KHE should be evaluated to determine if a complete resection without mutilation is possible. However, with the data collected it could not be clarified if there has to be a microscopically complete resection (R0) or if an R1 resection is enough.
If not resectable, we can confirm recent results that sirolimus is the most effective systemic treatment modality [21,22,23,24,25, 29,30,31]. However, this is in contrast to the metaanalysis of Liu et al. from 2016 [11, 28]. These authors concluded that weekly vincristine has higher response rates than corticosteroids, interferon, radiotherapy, embolization, aspirin/ticlopidin and sirolimus. In our analysis, with patients and data collected over the period from 1993 to December 2017, response of the KMP vs. regression of the tumor size was seen in 97 vs. 100% with sirolimus, in 65 vs. 61% with anti-platelet agents, in 47 vs. 39% with vincristine, in 44 vs. 43% with interferon alpha, in 43 vs. 43% with radiotherapy, in 29 vs. 28% with steroids, in 29 vs. 28% with embolization and in 28 vs. 28% with propranolol as single therapy and/or in combination with other agents. The therapeutic effect of sirolimus was, therefore, significantly greater than that of steroids with regard to treating KMP and reducing tumor size.
The low response rates (< 30%) for steroids, radiotherapy, propranolol and embolization lead to the conclusion that these treatments should no longer be used as first-line therapy, especially since some of them may cause long-term complications [33]. In addition, interferon alpha could also not be recommended due to its possible significant neurological side effects [34]. Our findings recommending sirolimus as the first-line therapy are also not in line with the consensus statement published 2013 from Drolet et al. [4]. They recommended vincristine 0.05 mg/kg/week and steroids (oral prednisolone 2 mg/kg/day or i.v. methylprednislone 1.6 mg/kg/day) for patients with KMP and oral prednisolone for patients with growing tumors or symptoms not related to KMP.
In the meta-analysis of Liu et al. [11], 37 patients from 3 studies [29, 32, 35] were treated with sirolimus with a response rate of 57% with a high heterogeneity. The average time until response was 25 days (range 8–64 days) [29]. Kai et al. reported that all 6 infants refractory to at least 2 other drugs responded to sirolimus [30]. In a prospective phase II trial with sirolimus 10/10 patients with KMP and 1 of 3 without KMP had a partial response of the hematologic disorder associated with a tumor reduction of > 20% [29]. In a multicenter, retrospective study looking at 52 patients with progressive KHE, sirolimus showed clinical improvement in 96% after 6 months [31]. Short-term steroids were given in addition in 21 patients with KMP, with no effect with respect to better tumor shrinkage. Importantly, there are no decisive data about the duration of sirolimus treatment. Currently, it is recommended that therapy is to be tailored to the individual response of the patient.
A phase II study randomizing sirolimus and vincristine started in 2017 and is still ongoing (Clinical Trials gov identifier NCT02110059). In contrast, the phase II study evaluating the safety and efficacy of sirolimus in complicated vascular anomalies (NCT00975819) completed recruitment in 2014; the results are expected in 2019. Those results are urgently needed.
Conclusions
In summary, we are able to show that sirolimus (starting with 0.8 mg/m2/dose every 12 hours, trough level 10–15 ng/mL) is the most effective therapy and would be an option for first-line treatment in patients with non-resectable and progressive tumors with or without KMP. The role of steroids in addition to sirolimus is not clear, but has at best an effect on KMP but not on reducing tumor size. Prospective data are urgently needed comparing the effects of vincristine/steroids with sirolimus monotherapy or sirolimus/short-term steroids.
References
Croteau SE, Liang MG, Kozakewich HP, Alomari AI, Fishman SJ, Mulliken JB, et al. Kaposiform hemangioendothelioma: atypical features and risks of Kasabach–Merritt phenomenon in 107 referrals. J Pediatr. 2012;162:142–7.
Deraedt K, Vander Poorten V, Van Geet C, Renard M, De Wever I, Sciot R. Multifocal kaposiform haemangioendothelioma. Virchows Arch. 2006;448:843–6.
O’Rafferty C, O’Regan GM, Irvine AD, Smith OP. Recent advances in the pathobiology and management of Kasabach-Merritt phenomenom. Br J Haematol. 2015;171:38–51.
Drolet BA, Trenor CC 3rd, Brandão LR, Chiu YE, Chun RH, Dasgupta R, et al. Consensus-derived practice standards plan for complicated kaposiform hemangioendothelioma. J Pediatr. 2013;163:285–91.
Sarkar M, Mulliken JB, Kozakewich HPW, Robertson RL, Burrows PE. Thrombocytopenic coagulopathy (Kasabach–Merritt phenomenon) is associated with Kaposiform hemangioendothelioma and not with common infantile hemangioma. Plast Reconstr Surg. 1997;100:1377–86.
Chu CY, Hsiao CH, Chiu HC. Transformation between kaposiform hemangioendothelioma and tufted angioma. Dermatology. 2003;206:334–7.
Zukerberg LR, Nickoloff BJ, Weiss SW. Kaposiform hemangioendothelioma of infancy and childhood. An aggressive neoplasm associated with Kasabach–Merritt syndrome and lymphangiomatosis. Am J Surg Pathol. 1993;17:321–8.
Lyons LL, North PE, Mac-Moune Lai F, Stoler MH, Folpe AL, Weiss SW. Kaposiform hemangioendothelioma: a study of 33 cases emphasizing its pathologic, immunophenotypic, and biologic uniqueness from juvenile hemangioma. Am J Surg Pathol. 2004;28:559–68.
Enjolras O, Mulliken JB, Wassef M, Frieden IJ, Rieu PN, Burrows PE, et al. Residual lesions after Kasabach–Merritt phenomenon in 41 patients. J Am Acad Dermatol. 2000;42:225–35.
Schaefer BA, Wang D, Merrow AC, Dickie BH, Adams DM. Long-term outcome for kaposiform hemangioendothelioma: a report of two cases. Pediatr Blood Cancer. 2017;64:284–6.
Liu XH, Li JY, Qu XH, Yan WL, Zhang, L, Yang C, et al. Treatment of kaposiform hemangioendothelioma and tufted angioma. Int J Cancer. 2016;139:1658–66.
Wang Z, Li K, Yao W, Dong K, Xiao X, Zheng S. Steroid-resistant kaposiform hemangioendothelioma: a retrospective study of 37 patients with vincristine and long-term follow-up. Pediatr Blood Cancer. 2015;62:577–80.
Fernandez-Pineda I, Lopez-Gutierrez JC, Chocarro G, Bernabeu-Wittel J, Ramirez-Villar GL. Long-term outcome of vincristine-aspirin-ticlopidine (VAT) therapy for vascular tumors associated with Kasabach–Merritt phenomenon. Pediatr Blood Cancer. 2013;60:1478–81.
Haisley-Royster C, Enjolras O, Frieden IJ, Garzon M, Lee M, Oranje A, et al. Kasabach–Merritt phenomenon: a retrospective study of treatment with vincristine. J Pediatr Hematol Oncol. 2002;24:459–62.
Fahrtash F, McCahon E, Arbuckle S. Successful treatment of kaposiform hemangioendothelioma and tufted angioma with vincristine. J Pediatr Hematol Oncol. 2010;32:506–10.
Shen W, Cui J, Chen J, Zou J, Ji Y, Chen H. Kasabach–Merritt syndrome with partial resection of tumor, reduction of tumor blood, and vincristine chemotherapy. J Craniofac Surg. 2010;21:215–6.
Hauer J, Graubner U, Konstantopoulos N, Schmidt S, Pfluger T, Schmid I. Effective treatment of kaposiform hemangioendotheliomas associated with Kasabach–Merritt phenomenon using four-drug regimen. Pediatr Blood Cancer. 2007;49:852–4.
Fuchimoto Y, Morikawa N, Kuroda T, Hirobe S, Kamagata S, Kumagai M, et al. Vincristine, actinomycin D, cyclophosphamide chemotherapy resolves Kasabach–Merritt syndrome resistant to conventional therapies. Pediatr Int. 2012;54:285–7.
Hu B, Lachman R, Phillips J, Peng SK, Sieger L. Kasabach–Merritt syndrome-associated kaposiform hemangioendothelioma successfully treated with cyclophosphamide, vincristine, and actinomycin D. J Pediatr Hematol Oncol. 1998;20:567–9.
Harper L, Michel JL, Enrolas O, Raynaud-Mounet N, Rivière JP, Heigele T, et al. Successful management of a retroperitoneal kaposiform hemangioendothelioma with Kasabach–Merritt phenomenon using alpha-interferon. Eur J Pediatr Surg. 2006;16:369–72.
Hammill AM, Wentzel M, Gupta A, Nelson S, Lucky A, Elluru R, et al. Sirolimus for the treatment of complicated vascular anomalies in children. Pediatr Blood Cancer. 2011;57:1018–24.
Uno T, Ito S, Nakzawa A, Miyazaki O, Mori T, Terashima K. Successful treatment of Kaposiform hemangioendothelioma with everolimus. Pediatr Blood Cancer. 2015;62:536–8.
Matsumoto H, Ozeki M, Hori T, Kanda K, Kawamoto N, Nagano A, et al. Successful everolimus treatment of kaposiform hemangioendothelioma with Kasabach–Merritt phenomenon: clinical efficacy and adverse effects of mTOR inhibitor therapy. J Pediatr Hematol Oncol. 2016;38:e322–5.
Blatt J, Stavas J, Moats-Staats B, Moats-Staats B, Woosley J, Morrell DS. Treatment of childhood kaposiform hemangioendothelioma with sirolimus. Pediatr Blood Cancer. 2010;55:1396–8.
Jahnel J, Lackner H, Reiterer F, Reiterer F, Urlesberger B, Urban C. Kaposiform hemangioendothelioma with Kasabach–Merritt phenomenon: from vincristine to sirolimus. Klin Padiatr. 2012;224:395–7.
Adams DM, Hammill AM, Mobberley-Schuman PS, Mobberley-Schuman PS, Trenor CC 3rd. Comment on: steroid-resistant kaposiform hemangioendothelioma: a retrospective study in 37 patients treated with vincristine and long-term follow-up. Pediatr Blood Cancer. 2015;62:2056.
Boccara O, Fraitag S, Lasne D, Fontaine J, Bughin V, Hamel-Teillac D, et al. Kaposiform haemangioendothelioma-sprectrum lesions with Kasabach–Merritt phenomenom: retrospective analysis and long-term outcome. Acta Derm Venereol. 2015;96:77–81.
Liu X, Li J, Qu X, Yan W, Zhang L, Zhang S, et al. Clinical outcomes for systemic corticosteroids versus vincristine in treating kaposiform hamangioendothelioma and tufted angioma. Medicine (Baltimore). 2016;95:e3431.
Adams DM, Trenor CC 3rd, Hammill AM, Vinks AA, Patel MN, Chaudry G, et al. Efficacy and safety of sirolimus in the treatment of complicated vascular anomalies. Pediatrics. 2016;137:e20153257.
Kai L, Wang Z, Yao W, Dong K, Xiao X. Sirolimus, a promising treatment for refractory kaposifrom hemangioendothelioma. J Cancer Res Clin Oncol. 2014;140:471–6.
Ji Y, Chen S, Xiang B, Li K, Xu Z, Yao W, et al. Sirolimus for the treatment of progressive kaposiform hemangioendothelioma: a multicenter retrospective study. Int J Cancer. 2017;141:848–55.
Gruman A, Liang MG, Mulliken JB, Fishman SJ, Burrows PE, Kozakewich HP, et al. Kaposiform hemangioendothelioma without Kasabach–Merritt phenomenon. J Am Acad Dermatol. 2005;52:616–22.
Mahajan P, Margolin J, Iacobas I. Kasabach–Merritt phenomenon: classic presentation and management options. Clin Med Insights Blood Disord. 2017;10:1179545X17699849.
Barlow CF, Priebe CJ, Mulliken JB, Barnes PD, Mac Donald D, Folkman J, et al. Spastic diplegia as a complication of interferon Alfa-2a treatment of hemangiomas of infancy. J Pediatr. 1998;132:527–30.
Tlougan BE, Lee MT, Drolet BA, Drolet BA, Frieden IJ, Adams DM, et al. Medical management of tumors associated with Kasabach–Merritt phenomenon: an expert survey. J Pediatr Hematol Oncol. 2013;35:618–22.
Funding
None
Author information
Authors and Affiliations
Contributions
IS wrote the manuscript, collected patient data and contributed to the analyses and interpretation of the data. AKK wrote the manuscript, collected patient data and contributed to the analyses and interpretation of the data. MSS collected patient data and contributed to the analyses and interpretation of the data. EK collected patient data and contributed to the analyses and interpretation of the data. RM wrote the manuscript, collected patient data and contributed to the analyses and interpretation of the data. BH wrote the manuscript, collected patient data and contributed to the analyses and interpretation of the data. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval
Not necessary since the article is a literature review and retrospective analysis.
Conflict of interest
None of the authors have any competing interests to disclose.
Rights and permissions
About this article

Cite this article
Schmid, I., Klenk, A.K., Sparber-Sauer, M. et al. Kaposiform hemangioendothelioma in children: a benign vascular tumor with multiple treatment options. World J Pediatr 14, 322–329 (2018). https://doi.org/10.1007/s12519-018-0171-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12519-018-0171-5