
Abstract
Attention deficit hyperactivity disorder (ADHD) is characterised by the typical behavioural core symptoms of inattentiveness, hyperactivity and impulsiveness. ADHD is a usually chronic health conditions, mostly diagnosed in childhood, creating a significant challenge for youth, their families and professionals who treat it. This disorder requires long-term treatments, including psychotherapeutic and pharmacological interventions, which in some cases may lead to adverse effects. Understanding the mechanism by which ADHD risk factors affect the biochemical processes in the human brain and consequentially the behaviour will help to identify novel targets for the development of therapeutics with less adverse results and better efficacy including higher responder rates. Although inflammatory responses in the brain have been recognised for years as critical in neurodegeneration and behaviour in a number of neurological and psychiatric disorders, their role for the development, treatment and prevention of ADHD has been so far largely overlooked, although historically, ADHD symptoms were initially observed in patients who survived an ONJ infection, i.e. inflammation. In this review, we discuss the interrelationship between different ADHD risk factors and inflammation with respect to the triggered molecular mechanisms and the contribution they are likely to have to this disorder. This paper provides a rationale for future studies on ADHD with an intent to inspiring the development of new agents for a more efficient management of this disorder.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Attention deficit hyperactivity disorder (ADHD) is one of the most common and highly heritable childhood-onset psychiatric disorders. A high degree of inattention with or without hyperactive-impulsive behaviour often results in impaired social and academic functioning. The estimated worldwide prevalence of ADHD is 5.3% in children and adolescents. The frequency of ADHD declines with age but can persist into adulthood, affecting between 2.5 and 4.4% of adults. At least 25% of adults with a history of hyperactivity are the biologic parent of a child with hyperactive symptoms (Sharp et al. 2009). At present, it is uncertain what the causes are for ADHD. Many studies suggest that a number of genes play a large role. However, it is likely that ADHD, like many other psychiatric disorders, does not represent a so-called nosological entity, but rather results from a combination of different factors, whose aetiopathogenetic role may differ among individual cases:
Genes
Studies estimate the mean heritability of ADHD to be 76%, indicating that ADHD is one of the most heritable psychiatric disorders. So far, more than thirty genes that are involved in neurodevelopment have been implicated in ADHD to some extent (Sharp et al. 2009). Deletion of some of these genes in animals (e.g. SNAP25, TACR1) results in phenotypes suggesting a strong association with some of the ADHD symptoms (Mill et al. 2004; Yan et al. 2010). However, none of the genes implicated have been shown to be solely responsible for the complex behavioural pattern in ADHD.
Environmental factors
Studies suggest a potential link between environmental factors such as cigarette smoking and alcohol use during pregnancy and ADHD in children (Linnet et al. 2003; Mick et al. 2002).
Brain injuries
Children who have suffered a brain injury may show some behaviours similar to those of ADHD (Kostrzewa et al. 2008). However, this is not likely to be a primary reason because only a small percentage of children with ADHD have suffered a traumatic brain injury.
Food additives
Recent research indicates a possible link between consumption of certain food additives like artificial colours or preservatives, and an increase in activity (McCann et al. 2007). However, the effect sizes found in this study are very small and therefore it is not clear if could contribute to the ADHD phenotype in a relevant extent. Alterations in the amounts of essential fatty acids from the omega-3 and omega-6 groups have been also found to be associated with ADHD (Raz and Gabis 2009). Further research is underway to confirm the findings and to learn more about the hyperactivity effect some food additives may have.
Atopic eczema
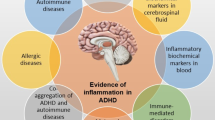
A number of recent studies demonstrated a significant association between atopic eczema (AE) and ADHD in children (Romanos et al. 2010; Schmitt et al. 2009, 2010). Considering that the AE is the most prevalent chronic inflammatory condition in children (Schmitt et al. 2009), this strongly suggests a role for inflammation in development of ADHD.
Here, we discuss the interrelationship between different ADHD risk factors and inflammation with respect to the triggered molecular mechanisms and their possible contribution to this disorder. More specifically, the mechanisms proposed here imply the role of inflammation in cell death, neuronal damage and brain development, which may constitute one possible mechanism among many others. Inflammation can play dual role in the brain. It can promote neurodegeneration or contribute to neuronal survival (Kolev et al. 2009). Inflammation has been also implemented as an important factor during brain development (reviewed by Jonakait 2007). ADHD is a neurodevelopmental disorder and therefore, the emphasis of this review is on the role of inflammatory responses on neurodevelopment and adult neurogenesis, which has been shown to affect ADHD phenotype.
Omega fatty acids and ADHD
Omega fatty acids are perhaps one of the most extensively studied biomarkers for ADHD. There are three major dietary omega-3 (n-3) fatty acids: α-linolenic acid (ALA) (C18:3 isomer), eicosapentaenoic acid (EPA) (C20:5 isomer) and docosahexaenoic acid (DHA) (C22:6 isomer). In the context of the modern human lifestyle and diet, the eicosanoid metabolites of EPA are crucial to provide anti-inflammatory effects by balancing the potentially pro-inflammatory eicosanoid metabolites of the omega-6 (n-6) arachidonic acid (ARA). It is important to stress that ARA metabolites are not innately dangerous to health. The organism requires ARA for systemic homoeostasis at higher nutritional levels than n-3 fatty acids. n-6/n-3 ratios of around 4/1 are considered to be optimal. In a study, ADHD was associated with low red blood cell DHA and high n-6 fatty acid levels (Riediger et al. 2009). This association and the role of each of the fatty acids in modulating the inflammatory responses suggest that increased inflammation in ADHD as a result of altered ratio n-6/n-3 may be a risk factor for this disorder. However, so far there are no studies that address the role of different ratios between n-6 and n-3 for the inflammatory responses in the brain, which may elucidate the mechanisms by which the altered levels of expression of these fatty acids affect the behaviour. Recently, the pro-apoptotic role of ARA was demonstrated, while DHA was shown to inhibit this process (King et al. 2006). These findings correlate very well with previous observations that neurobehavioural deficits are associated with apoptotic neurodegeneration and vulnerability for ADHD (Fredriksson and Archer 2004) and with the finding of decreased DHA and increased ARA in patients suffering from this disorder.
Based on the disbalanced n-6/n-3 ratio in individuals with ADHD, a number of clinical studies and trials on the use of fatty acids for treatment in ADHD have been carried out (reviewed by (Raz and Gabis 2009). Randomised controlled trials, however, have generally been unsuccessful in demonstrating any behavioural treatment effects. It can be speculated that a reason for this is the higher activity of enzymes involved in the fatty acids catabolism in ADHD patients. Essential fatty acids are efficiently transported via the blood brain barrier (Spector 1988) and therefore their uptake by the brain should be well possible. However, the essential fatty acids and glutathione deficiency observed in ADHD (Bradstreet et al. 2010; Riediger et al. 2009) can cause malfunction of the blood brain barrier.
Another important mechanism by which altered ratios of essential fatty acids in ADHD may affect patients’ behaviour is their involvement in building the lipid raft in neurons and other cell types. DHA has been shown to be of particular importance for growth and function of nervous tissue. Reduced DHA is associated with impairments in cognitive and behavioural performance, effects which are particularly important during brain development (Fenton et al. 2001). Recent studies suggest participation of DHA in neurogenesis (Coti Bertrand et al. 2006; Kawakita et al. 2006), neurotransmission (Chalon 2006) and protection against oxidative stress (Bazan 2006). These functions relate to the roles of DHA within the hydrophobic core of neural membranes, such as conferring a high degree of flexibility and direct interaction with membrane proteins, thus impacting speed of signal transduction, neurotransmission and formation of lipid rafts (Chalon 2006; Grossfield et al. 2006; Stillwell et al. 2005). DHA has been shown to modulate the activity of major signalling pathways in neurodevelopment controlled by protein kinase A (PKA) (Tai et al. 2009) and protein kinase C (PKC) (Massaro et al. 2006), which orchestrate major pathways controlling inflammatory responses in the brain.
Brain injury, protein kinases and ADHD
The critical role of protein kinases in brain development has been known and extensively investigated for decades. Surprisingly, the role of protein kinases for the development of ADHD has not been studied comprehensively and only recently researchers started to address this issue. Haplotype-based association analysis for two single nucleotide polymorphisms (SNPs) located in Intron 3 (rs402691) and Exon 6 (rs3745406) of the PKC-γ (PRKCG) gene indicated a significant overall association of this locus with the ADHD hyperactive subscale scores in Caucasians, suggesting a relation between impulsive behaviours and the PRKCG gene (Schlaepfer et al. 2007). Unfortunately, no follow-up studies have been carried out to address in greater details the functional role of these ADHD-associated SNPs in the PRKCG locus. Another recent study found that PKA inhibition within the medial prefrontal cortex of rats produces inattention and hyperactivity, and thus might be useful in modelling human attention disorders (Paine et al. 2009). Considering the implication of these two protein kinases in ADHD, it is important to know the effect of each one in inflammatory responses in the brain and the alterations in their activity upon brain injury, a possible risk factor for ADHD.
PKA controls the expression of some key cytokines, such as IL-6 (Bergamaschi et al. 2006). IL-6 is an interleukin that acts as both a pro-inflammatory and anti-inflammatory cytokine. IL-6 role as an anti-inflammatory cytokine is mediated through its inhibitory effects on TNF-α and IL-1, and activation of IL-1ra and IL-10. Upon brain injury, however, the activity of PKA is attenuated (Atkins et al. 2007) and production of IL-6 increased (Shohami et al. 1994; Taupin et al. 1993). It is now established that IL-6 inhibits neurogenesis in the hippocampus through blockade of the differentiation of neural progenitor cells into neurons (Monje et al. 2003), which may create serious problem in developing brains and could result in an increased risk from ADHD. During brain injury, extracellular adenosine and glutamate levels increase rapidly and dramatically, which redirects the adenosine A2AR receptor signalling from the PKA to the PKC pathway, resulting in a switch in A2AR effects from anti-inflammatory to pro-inflammatory (Dai et al. 2010). PKA has an anti-apoptotic effect on neurons (Kholodenko et al. 2007) and its inhibition therefore results in increased neuronal death which is a major feature in animal models of ADHD (Fredriksson and Archer 2004). On the other hand, inhibited PKA results in the dephosphorylation of C3, a central component of the complement system, allowing enhanced activation of complement cascade (Forsberg et al. 1990). It is now established that different types of brain cells can locally synthesise a fully functional complement system without the need of transporting these proteins via the blood–brain barrier. Both glia and nerve cells in the CNS can synthesise immunoglobulins (Asghar and Pasch 2000) and complement components (Barnum 1995; Silva et al. 1985). Brain cells can express all of the complement components and should thus be considered as an important source of complement (Emmerling et al. 2000). There is recent evidence that besides the generation of inflammatory responses by the complement system, it may also play a neuroprotective role (reviewed by (Donev et al. 2009). The most convincing data for the protective role of components of the complement cascade demonstrate that the production of C5a results in an activation of the neuroprotective p38 mitogen–activated protein kinases (p38 MAPK) and reduced neuronal apoptosis (Osaka et al. 1999). Furthermore, animals genetically deficient in the complement component C5 were found to be more susceptible to hippocampal excitotoxic lesions (Pasinetti 1996). These findings suggest that inflammatory responses in the brain can be also beneficial in ADHD by generating non-inflammatory molecules which at least in part could moderate the negative effects. This issue, however, has not been discussed so far. Our unpublished data suggest that augmenting p38 MAPK by early stages of activation of complement system is protective against ADHD pathology, particularly during childhood when the brain still develops (Toga et al. 2006).
Smoking, inflammation and ADHD
It is believed that in general smoking is a risk factor for ADHD, although some contradictive findings suggesting that the previously observed association between maternal smoking and ADHD might represent an inherited/familial confound have recently been published (Thapar et al. 2009). Comprehensive analysis of the published in the literature data shows that it promotes rather a complex response, which is likely to have dual effects on the brain. Cigarette smoke has been reported to have inflammatory effects such as increasing peripheral leucocyte counts, and recruitment of polymorphonuclear cells, monocytes and macrophages (Costenbader and Karlson 2006). It has also been shown to increase levels of C-reactive protein (CRP), which is an acute phase inflammation marker (Hakobyan et al. 2008). It is likely that such an increase would have a dual role as a risk factor for ADHD. CRP is known to induce apoptosis in different cell types (Nabata et al. 2008) which should contribute towards brain dysfunction. On the other hand, however, CRP binds to apoptotic cells, protects the cells from assembly of the terminal complement components, i.e. form membrane attack complex to which neurons are very vulnerable (Kolev et al. 2010), and sustains an anti-inflammatory innate immune response (Gershov et al. 2000). CRP, however, is only moderately expressed in the brain. In recent years, an abundant and brain-specific protein S100B was found to have similar functions in the central nervous system (CNS) as the CRP in the blood stream and tissues exposed to it (Sen and Belli 2007). S100B exerts both intracellular and extracellular functions. Intracellular S100B acts as a stimulator of cell proliferation and migration and an inhibitor of apoptosis and differentiation, which might have important implications during brain development and repair, activation of astrocytes in the course of brain damage and neurodegenerative processes (reviewed by (Donato et al. 2009). The effects caused by the intracellular S100B suggest that its increased expression as a result of smoking (Morbini et al. 2006) may have both beneficial (anti-apoptotic) and negative (dedifferentiation) effects within the pathophysiological cascade of ADHD. The dual effect of smoking-increased extracellular S100B, mediated via the S100B receptor RAGE (receptor for advanced glycation and products), is very similar to that observed for the intracellular protein.
Cigarette smoke contains various free radicals including nitric oxide (NO) (Church and Pryor 1985). Increased NO plasma levels in smokers have also been reported in several studies (Miller et al. 1998; Sarkar et al. 1999; Zhou et al. 2000), although not confirmed in all studies (Node et al. 1997). NO is an important neurotransmitter and signalling molecule that acts in many tissues to regulate a diverse range of physiological processes including vasodilation, neuronal function, inflammation and immune function (Clancy et al. 1998; Vincent 2010). NO has also been reported to cause axonal degeneration or block axonal conduction especially in physiologically active or demyelinated axons (Kapoor et al. 2003; Redford et al. 1997; Smith et al. 2001). iNOS is the high-output isoform of the nitric oxide synthase (NOS) (Encinas et al. 2005). iNOS has been shown to be expressed in multiple cell types, including astrocytes, and has been suggested to contribute to vasodilation and damage to the blood–brain barrier besides its immunoregulatory role (Liu et al. 2001). Furthermore, NO has also been demonstrated to be involved in the regulation of apoptosis (Kaur and Ling 2008). The effects of apoptosis vary depending upon the dose of NO and the cell type used and has been shown to be able to both induce apoptosis and to protect from apoptosis in different cell types. In neurons, however, it seems that high NO levels have a pro-apoptotic effect mediated via the neuronal NO synthase (García-Nogales et al. 2003), which further contributes towards classifying NO as a potential risk factor for ADHD. Studies from the peripheral nervous system indicate a key role for voltage-sensitive calcium channels in regulating NO synthase activity. A similar mechanism may also be essential in central neurons, and it remains an important task to identify the precise sources of calcium-regulated NO production in neurons. Also, although cGMP production appears to mediate the physiological signalling by NO, the specific roles of cGMP-dependent ion channels, protein kinases and phosphodiesterases in mediating NO action remain to be determined (Vincent 2010). Considering the above functions of NO, it is not unexpected that a functional promoter dinucleotide repeat length variation of the neuronal nitric oxide synthase gene (NOS1 Ex1f-VNTR) was found to be associated with impulsivity-related behavioural phenotypes, including ADHD (Reif et al. 2009; Retz et al. 2010).
Another complex chain of events in the CNS triggered by smoking starts with the up-regulated expression of the serum intercellular adhesion molecule 1 (ICAM-1) and E-selectin (Costenbader and Karlson 2006). The expression of these two endothelial adhesion molecules in the CNS facilitates the entry of T cells into the brain (Noseworthy et al. 2000). S100B, also overexpressed by smoking, can activate T cells via RAGE, resulting in their activation and the generation of pro-inflammatory cytokines (e.g. IFNγ) and adhesion molecules (Hofmann et al. 1999; Yan et al. 2003). Activated T cells, some of which express IL-17 promoting blood–brain barrier disruption (Huppert et al. 2010), could infiltrate via the blood–brain barrier contributing to the increased apoptosis observed in ADHD brains. It is important to stress that IFNγ secreted by T cells does not alter the activity of p38 MAPK (Smith et al. 2007) and therefore, no beneficial effect from an increased presence of T cells would be expected.
Given the data presented up to here, it seems that smoking could be a very serious risk factor for ADHD. However, some other studies demonstrate a rather positive consequence from nicotine for modifying symptoms related to ADHD. Much of the scientific literature examining the causes of ADHD points to the neurotransmitters dopamine and norepinephrine and to frontal brain regions. Those same brain regions also contain significant numbers of nicotinic receptors when a person smokes. The receptors have other functions, too, but among others they are thought to be involved in nicotine’s addictive properties and in the “positive effects” it provides. Some studies have found that nicotine can increase attention span, in both animals and adults with normal attention and in those with ADHD (Min et al. 2001; Rushforth et al. 2010). It appears that one possible reason why many of ADHD children smoke is that they are “self” treating their attention problems in a very specific, pharmacological way. However, nicotine has too many other side effects, as described above, and therefore cannot be used therapeutically. A recent study showed that activity levels of two major components of cAMP signalling, cAMP-dependent PKA and adenylate cyclase, were elevated in the nucleus accumbens of smokers and in the ventral midbrain dopaminergic region of smokers as well as former smokers (Hope et al. 2007). PKA expression levels were also found to be over-expressed in smokers and former smokers. These findings suggest that smoking-induced neuroadaptations in the brain can persist for significant periods in former smokers. This study together with the finding that PKA inhibition within the medial prefrontal cortex of rats produces inattention and hyperactivity (Paine et al. 2009) implies that the increased expression and activity of PKA are likely to account, at least in part, for the “beneficial” effect of nicotine on attention.
Alcohol, inflammation and ADHD
Alcohol and its metabolism can modulate the cellular response to microflora-derived lipopolysaccharide (LPS), a strong inducer of immune responses and activator of numerous transcription factors, in the liver and independently generate endogenous inflammatory inducers. Thus, both alcohol and LPS act simultaneously to influence the inflammatory response in the liver and other organs, including the brain (Wang et al. 2010). Long-term alcohol exposure impairs the liver’s ability to produce pro-inflammatory, and more significantly, anti-inflammatory cytokines. Circulating monocytes in patients suffering from alcoholism show reduced IL-10 production in response to LPS (Hill et al. 2002; Le Moine et al. 1995; McClain et al. 2002). The reduced IL-10 production in alcoholic patients is associated with an increased production of TNF-α by circulating monocytes (McClain and Cohen 1989). The reduction of circulating IL-10 together with increased IL-6 levels is likely to account for a direct role in the appearance of pro-inflammatory Th17 cells in circulation (Lemmers et al. 2009) and their increased infiltration in the brain (Huppert et al. 2010). Thus, chronic alcohol exposure creates an imbalance in the cytokine environment favouring systemic inflammatory conditions that can cause cell injury. The central pro-inflammatory cytokines induced by alcohol, in particular IL-6, inhibit neurogenesis in the hippocampus through blockade of the differentiation of neural progenitor cells into neurons (Monje et al. 2003). Recent study underscored the involvement of adult hippocampal neurogenesis in psychiatric disorders, including ADHD (Lagace et al. 2007). Investigators showed that methylphenidate, a drug use for treatment of ADHD, administered during juvenile development produces long-term behavioural changes in adult animals.
Even in the absence of specific neurological complications, excessive drinking can lead to regional structural brain damage and cognitive dysfunction (Harper and Matsumoto 2005), neuronal death and inhibition of neurogenesis (Crews and Nixon 2009), resulting in a reduction in brain volume, including white matter (Harper 1998). Recently, a mechanism by which alcohol causes neurodegeneration was proposed. It was shown that TNF-α and the pro-inflammatory chemokine MCP-1 are induced in the brains of mice treated intragastrically with alcohol (Qin et al. 2008). Separately, it has been suggested that alcohol causes neurodegeneration by reducing the activity of the pro-neuronal survival transcription factor CREB and by activating the inflammation-promoting transcription factor NF-κB (Crews and Nixon 2009). Previous studies however gave conflicting data demonstrating that in vivo exposure to alcohol increases GABA release via up-regulation of the cAMP/PKA pathway (Bonci and Williams 1997; Melis et al. 2002). This increase of the PKA activity implies that alcohol should enhance the activity of CREB. Obviously, more studies are necessary to clarify the exact processes which lead to neurodegeneration by the alcohol. In summary, alcohol-promoted inflammation seems to contribute to the ADHD development via multiple mechanisms such as inhibition of neurogenesis and the pro-neuronal survival transcription factor CREB, increased activity of the inflammation-promoting NF-κB and T cells infiltration via the blood–brain barrier.
Atopic eczema, inflammation and ADHD
A number of recent studies indicated that comorbidities of eczema are not limited to allergic reactions, but also that children and adolescents with AE are diagnosed with mental health problems such as ADHD significantly more frequent than children with no history of eczema (Romanos et al. 2010; Schmitt et al. 2009, 2010). This intriguing finding for the association between AE and ADHD could be explained by the fact that AE is the most prevalent chronic inflammatory condition in children. An elevated pro-inflammatory Th17 cell frequency has been found in patients with AE (Aerts et al. 2010). These cells secret IL-17 that induces blood–brain barrier disruption and allows their increased infiltration in the brain (Huppert et al. 2010). There are now compelling data to suggest that chronic cytokine activation may contribute substantially to the phenotype and effector function of T cells. The infiltrated and activated T cells can contribute to the increased apoptosis found in ADHD brains, a feature of ADHD (Fredriksson and Archer 2004).
Genetics of ADHD and inflammation signalling
A number of neurodevelopmental genes and SNPs/deletions within them have been found to be possibly associated with ADHD (Sharp et al. 2009). Here, we discuss only those which alter inflammatory signalling in the brain.
Recently, it was discovered that genomic region at 16p11.2 is subject to recurrent copy number variations in patients with autism spectrum disorder and ADHD symptoms (Shinawi et al. 2009; Shiow et al. 2009). Patients with a duplicated 16p11.2 region had microcephaly and ADHD. This duplicated chromosomal region encompasses 25 genes, including MAPK3/ERK1. MAPK3 transduces signals from growth factors and is a key regulator of differentiation and proliferation in many cell types, including neurons. The inhibition of MAPK3 is thought to be beneficial in a number of experimental models of neurodegenerative and chronic inflammatory disease. The MAPK3 signalling pathways have been found to be involved in microglial/macrophage activation (Bhat et al. 1998; Choi et al. 2003; Tikka et al. 2001). Previous studies show that the expression of activated MAPK3 in microglia/macrophages may play a key role in the production of CNS iNOS, cyclooxygenase-2 (COX-2), several pro-inflammatory cytokines (e.g. IL-1β, IL-6) and TNF-α (Combs et al. 2001; Conrad et al. 2000). Therefore, the duplication of 16p11.2 found to possibly be associated with ADHD is likely to contribute to a damage of the blood–brain barrier and a pro-apoptotic effect mediated via iNOS and pro-inflammatory cytokines (García-Nogales et al. 2003).
Another recent genetic study using single-marker and haplotype-based analyses provided evidence of a possible association between the Ciliary Neurotrophic Factor Receptor (CNTFR) and both adulthood and childhood ADHD and also suggested a childhood-specific contribution of neurotrophin 3 (NTF3) and neurotrophic tyrosine kinase receptor type 2 (TYK2) to ADHD (Ribasés et al. 2008). Neurotrophins are a family of proteins which are best characterised by their modulation of survival, differentiation and apoptosis of cells in the nervous system. This family includes nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), NTF3 and neurotrophins 4/5 (NTF4/5). Neurotrophins signal through the high-affinity tyrosine kinase receptors (TYK1/2/3), and the low affinity receptor p75NTR, a member of the tumour necrosis factor receptor family (Barbacid et al. 1991; Levi-Montalcini 1987). Unfortunately, most of the identified polymorphisms in the genes associated with ADHD have not been assessed for their functional effect on protein expression and/or activity. Thus, it is not possible to accurately understand the processes which may occur as a result of these polymorphisms. However, it is expected that the SNPs found in TYK2 would result in abnormal responses to inflammatory challenges in a variety of cells. TYK2 is a tyrosine kinase that acts upstream of STAT3 to induce caspase-3-dependent neuronal death (Wan et al. 2010). Therefore, the SNPs in the TYK2 would probably alter apoptosis levels. TYK2 is a receptor for the BDNF whose SNPs were to some degree associated with ADHD (Lasky-Su et al. 2008). Exposure to BDNF results in increased IL-6 gene and protein expression which is required for the activation of ERK and p38 MAPK signalling pathways (Rezaee et al. 2010). SNPs in BDNF and/or TYK2 will lead to dysbalanced signalling which is difficult to be accurately predicted with the information presently available. The other neurotrophic factor, CNTF, in whose receptor SNPs associated with ADHD were found (Ribasés et al. 2008), inhibits the production of TNF-α and therefore has a neuroprotective role. There are only a few studies investigating the role of CNTF on production of anti-inflammatory cytokines (e.g. IL-10) and they are rather inconclusive. Moreover, this issue has not been addressed in neurons in vivo. However, it is likely that the SNPs in the CNTFR found in ADHD patients would diminish the neuroprotective effect of CNTF. Our recent study showed that a SNP within NTF3 (rs6332) has a trend towards an association with ADHD (Conner et al. 2008). Although rs6332 is a silent Pro–Pro genetic variation, suggesting limited biological relevance of the mutation itself, this nucleotide replacement may have an effect on expression levels of the NTF3 which would have an impact on the CNTFR-controlled downstream signalling events and cytokine production.
IL-1, its antagonist, IL-1Ra and IL-1 receptors are all present in the brain, and IL-1 has been shown to influence both dopaminergic and noradrenergic function. The IL-1 superfamily consists of two members—IL-1α and IL-1β. Both are pro-inflammatory cytokines involved in immune defence against infection. The IL-1RA is a molecule that competes for receptor binding with IL-1α and IL-1β, blocking their role in immune activation. Polymorphisms in IL-1Ra gene, IL1RN, have been directly associated with ADHD (Segman et al. 2002). For the inheritance of alleles of an intronic 86-bp VNTR polymorphism, investigators reported significant evidence for biased transmission of the 4-repeat allele. This finding demonstrated the importance of inflammatory responses in the brain for this disorder. Again, no functional consequences from the polymorphisms that were found as risk factors for ADHD have been determined. Another study that attempted to replicate these findings, however, failed to find evidence for an association of this IL1RN polymorphism with ADHD (Misener et al. 2004). The lack of sufficient data on the role of inflammation in ADHD justifies further thorough investigations in this direction.
We have been able to generate unpublished data clearly demonstrating an association of SNPs in genes regulating the activity of complement system with ADHD. The SNPs found in patients result in inhibition of C5 cleavage by the complement cascade, which decreases the amount of the generated C5a, respectively. The lack of C5a, therefore, will act as a pro-apoptotic factor for neurons via maintaining low activity of the p38 MAPK, resulting in neuronal death.
Concluding remarks
It is becoming increasingly clear that inflammatory responses in the brain as a result of different environmental and genetics risk factors play a significant role in human behaviour in general and, in particular, in ADHD. Environmental factors have been shown to moderate the effects of genes on psychiatric disorders such as ADHD (Reif et al. 2007). Studies of gene–environment interactions in ADHD, that are still poorly understood, are essential to predict the disease-risk in asymptomatic individuals. Although the majority of data in the literature lead us towards the conclusion that inflammation in the brain seems to promote neurodegeneration and therefore should be a risk factor for ADHD, there are a rising number of examples that support the beneficial role of certain levels of inflammation for neuronal survival and function. Due to the exceptionally complex nature of the inflammatory network and the interrelationship between different inflammatory pathways, it is very difficult to predict the exact outcome and level of hazard each ADHD risk factor may have on different individuals. Moreover, it is most likely that the inflammatory response in the brain of each individual will depend on a very unique set of genetic factors. In order to obtain a more realistic and complex picture on the effect of risk factors on inflammatory processes in the brain, further studies that integrate genetics, functional genomics, proteomics, neuroimmunology and behavioural biology are necessary. Such integrative studies will provide a dynamic interpretation of biological processes in ADHD and new targets for the development of therapeutics for more efficient treatments for ADHD with less adverse effects.
References
Aerts N, De Knop K, Leysen J, Ebo D, Bridts C, Weyler J, Stevens W, De Clerck L (2010) Increased IL-17 production by peripheral T helper cells after tumour necrosis factor blockade in rheumatoid arthritis is accompanied by inhibition of migration-associated chemokine receptor expression. Rheumatology (in press)
Asghar S, Pasch M (2000) Therapeutic inhibition of the complement system. Y2K update. Front Biosci 5:E63–E81
Atkins C, Oliva AJ, Alonso O, Pearse D, Bramlett H, Dietrich W (2007) Modulation of the cAMP signaling pathway after traumatic brain injury. Exp Neurol 208:145–158
Barbacid M, Lamballe F, Pulido D, Klein R (1991) The trk family of tyrosine protein kinase receptors. Biochim Biophys Acta 1072:115–127
Barnum S (1995) Complement biosynthesis in the central nervous system. Crit Rev Oral Biol Med 6:132–146
Bazan N (2006) Cell survival matters: docosahexaenoic acid signaling, neuroprotection and photoreceptors. Trends Neurosci 29:263–271
Bergamaschi A, Corsi M, Garnier M (2006) Synergistic effects of cAMP-dependent signalling pathways and IL-1 on IL-6 production by H19-7/IGF-IR neuronal cells. Cell Signal 18:1679–1684
Bhat N, Zhang P, Lee J, Hogan E (1998) Extracellular signal-regulated kinase and p38 subgroups of mitogen-activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-alpha gene expression in endotoxin-stimulated primary glial cultures. J Neurosci 18:1633–1641
Bonci A, Williams J (1997) Increased probability of GABA release during withdrawal from morphine. J Neurosci 17:796–803
Bradstreet J, Smith S, Baral M, Rossignol D (2010) Biomarker-guided interventions of clinically relevant conditions associated with autism spectrum disorders and attention deficit hyperactivity disorder. Altern Med Rev 15:15–32
Chalon S (2006) Omega-3 fatty acids and monoamine neurotransmission. Prostaglandins Leukot Essent Fatty Acids 75:259–269
Choi S, Joe E, Kim S, Jin B (2003) Thrombin-induced microglial activation produces degeneration of nigral dopaminergic neurons in vivo. J Neurosci 23:5877–5886
Church DF, Pryor WA (1985) Free-radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect 64:111–126
Clancy RM, Amin AR, Abramson SB (1998) The role of nitric oxide in inflammation and immunity. Arthritis Rheum 41:1141–1151
Combs C, Karlo J, Kao S, Landreth G (2001) beta-Amyloid stimulation of microglia and monocytes results in TNFalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci 21:1179–1188
Conner A, Kissling C, Hodges E, Hünnerkopf R, Clement R, Dudley E, Freitag C, Rösler M, Retz W, Thome J (2008) Neurotrophic factor-related gene polymorphisms and adult attention deficit hyperactivity disorder (ADHD) score in a high-risk male population. Am J Med Genet B Neuropsychiatr Genet 147B:1476–1480
Conrad C, Marshall P, Talent J, Malakowsky C, Choi J, Gracy R (2000) Oxidized proteins in Alzheimer’s plasma. Biochem Biophys Res Commun 275:678–681
Costenbader KH, Karlson EW (2006) Cigarette smoking and autoimmune disease: what can we learn from epidemiology? Lupus 15:737–745
Coti Bertrand P, O’Kusky J, Innis S (2006) Maternal dietary (n-3) fatty acid deficiency alters neurogenesis in the embryonic rat brain. J Nutr 136:1570–1575
Crews F, Nixon K (2009) Mechanisms of neurodegeneration and regeneration in alcoholism. Alcohol Alcohol 44:115–127
Dai S, Zhou Y, Li W, An J, Li P, Yang N, Chen X, Xiong R, Liu P, Zhao Y, Shen H, Zhu P, Chen J (2010) Local glutamate level dictates adenosine A2A receptor regulation of neuroinflammation and traumatic brain injury. J Neurosci 30:5802–5810
Donato R, Sorci G, Riuzzi F, Arcuri C, Bianchi R, Brozzi F, Tubaro C, Giambanco I (2009) S100B’s double life: intracellular regulator and extracellular signal. Biochim Biophys Acta 1793:1008–1022
Donev R, Kolev M, Millet B, Thome J (2009) Neuronal death in Alzheimer’s disease and therapeutic opportunities. J Cell Mol Med 13:4329–4348
Emmerling M, Watson M, Raby C, Spiegel K (2000) The role of complement in Alzheimer’s disease pathology. Biochim Biophys Acta 1502:158–171
Encinas JM, Manganas L, Enikolopov G (2005) Nitric oxide and multiple sclerosis. Curr Neurol Neurosci Rep 5:232–238
Fenton W, Dickerson F, Boronow J, Hibbeln J, Knable M (2001) A placebo-controlled trial of omega-3 fatty acid (ethyl eicosapentaenoic acid) supplementation for residual symptoms and cognitive impairment in schizophrenia. Am J Psychiatry 158:2071–2074
Forsberg P, Martin S, Nilsson B, Ekman P, Nilsson U, Engström L (1990) In vitro phosphorylation of human complement factor C3 by protein kinase A and protein kinase C. Effects on the classical and alternative pathways. J Biol Chem 265:2941–2946
Fredriksson A, Archer T (2004) Neurobehavioural deficits associated with apoptotic neurodegeneration and vulnerability for ADHD. Neurotox Res 6:435–456
García-Nogales P, Almeida A, Bolaños J (2003) Peroxynitrite protects neurons against nitric oxide-mediated apoptosis. A key role for glucose-6-phosphate dehydrogenase activity in neuroprotection. J Biol Chem 278:864–874
Gershov D, Kim S, Brot N, Elkon K (2000) C-reactive protein binds to apoptotic cells, protects the cells from assembly of the terminal complement components, and sustains an antiinflammatory innate immune response: implications for systemic autoimmunity. J Exp Med 192:1353–1364
Grossfield A, Feller S, Pitman M (2006) A role for direct interactions in the modulation of rhodopsin by omega-3 polyunsaturated lipids. Proc Natl Acad Sci USA 103:4888–4893
Hakobyan S, Harris C, van den Berg C, Fernandez-Alonso M, de Jorge E, de Cordoba S, Rivas G, Mangione P, Pepys M, Morgan B (2008) Complement factor H binds to denatured rather than to native pentameric C-reactive protein. J Biol Chem 283:30451–30460
Harper C (1998) The neuropathology of alcohol-specific brain damage, or does alcohol damage the brain? J Neuropathol Exp Neurol 57:101–110
Harper C, Matsumoto I (2005) Ethanol and brain damage. Curr Opin Pharmacol 5:73–78
Hill D, D’Souza N, Lee E, Burikhanov R, Deaciuc I, de Villiers W (2002) A role for interleukin-10 in alcohol-induced liver sensitization to bacterial lipopolysaccharide. Alcohol Clin Exp Res 26:74–82
Hofmann M, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath M, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt A (1999) RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 97:889–901
Hope B, Nagarkar D, Leonard S, Wise R (2007) Long-term upregulation of protein kinase A and adenylate cyclase levels in human smokers. J Neurosci 27:1964–1972
Huppert J, Closhen D, Croxford A, White R, Kulig P, Pietrowski E, Bechmann I, Becher B, Luhmann H, Waisman A, Kuhlmann C (2010) Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J 24:1023–1034
Jonakait G (2007) The effects of maternal inflammation on neuronal development: possible mechanisms. Int J Dev Neurosci 25:415–425
Kapoor R, Davies M, Blaker PA, Hall SM, Smith KJ (2003) Blockers of sodium and calcium entry protect axons from nitric oxide-mediated degeneration. Ann Neurol 53:174–180
Kaur C, Ling E (2008) Antioxidants and neuroprotection in the adult and developing central nervous system. Curr Med Chem 15:3068–3080
Kawakita E, Hashimoto M, Shido O (2006) Docosahexaenoic acid promotes neurogenesis in vitro and in vivo. Neuroscience 139:991–997
Kholodenko R, Kholodenko I, Sorokin V, Tolmazova A, Sazonova O, Buzdin A (2007) Anti-apoptotic effect of retinoic acid on retinal progenitor cells mediated by a protein kinase A-dependent mechanism. Cell Res 17:151–162
King V, Huang W, Dyall S, Curran O, Priestley J, Michael-Titus A (2006) Omega-3 fatty acids improve recovery, whereas omega-6 fatty acids worsen outcome, after spinal cord injury in the adult rat. J Neurosci 26:4672–4680
Kolev M, Ruseva M, Harris C, Morgan B, Donev R (2009) Implication of complement system and its regulators in Alzheimer’s disease. Curr Neuropharmacol 7:1–8
Kolev M, Tediose T, Sivasankar B, Harris C, Thome J, Morgan B, Donev R (2010) Upregulating CD59: a new strategy for protection of neurons from complement-mediated degeneration. Pharmacogenomics J 10:12–19
Kostrzewa R, Kostrzewa J, Kostrzewa R, Nowak P, Brus R (2008) Pharmacological models of ADHD. J Neural Transm 115:287–298
Lagace D, Noonan M, Eisch A (2007) Hippocampal neurogenesis: a matter of survival. Am J Psychiatry 164:205
Lasky-Su J, Neale B, Franke B, Anney R, Zhou K, Maller J, Vasquez A, Chen W, Asherson P, Buitelaar J, Banaschewski T, Ebstein R, Gill M, Miranda A, Mulas F, Oades R, Roeyers H, Rothenberger A, Sergeant J, Sonuga-Barke E, Steinhausen H, Taylor E, Daly M, Laird N, Lange C, Faraone S (2008) Genome-wide association scan of quantitative traits for attention deficit hyperactivity disorder identifies novel associations and confirms candidate gene associations. Am J Med Genet B Neuropsychiatr Genet 147B:1345–1354
Le Moine O, Marchant A, De Groote D, Azar C, Goldman M, Devière J (1995) Role of defective monocyte interleukin-10 release in tumor necrosis factor-alpha overproduction in alcoholics cirrhosis. Hepatology 22:1436–1439
Lemmers A, Moreno C, Gustot T, Maréchal R, Degré D, Demetter P, de Nadai P, Geerts A, Quertinmont E, Vercruysse V, Le Moine O, Devière J (2009) The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology 49:646–657
Levi-Montalcini R (1987) The nerve growth factor 35 years later. Science 237:1154–1162
Linnet K, Dalsgaard S, Obel C, Wisborg K, Henriksen T, Rodriguez A, Kotimaa A, Moilanen I, Thomsen P, Olsen J, Jarvelin M (2003) Maternal lifestyle factors in pregnancy risk of attention deficit hyperactivity disorder and associated behaviors: review of the current evidence. Am J Psychiatry 160:1028–1040
Liu JS, Zhao ML, Brosnan CF, Lee SC (2001) Expression of inducible nitric oxide synthase and nitrotyrosine in multiple sclerosis lesions. Am J Pathol 158:2057–2066
Massaro M, Habib A, Lubrano L, Del Turco S, Lazzerini G, Bourcier T, Weksler B, De Caterina R (2006) The omega-3 fatty acid docosahexaenoate attenuates endothelial cyclooxygenase-2 induction through both NADP(H) oxidase and PKC epsilon inhibition. Proc Natl Acad Sci USA 103:15184–15189
McCann D, Barrett A, Cooper A, Crumpler D, Dalen L, Grimshaw K, Kitchin E, Lok K, Porteous L, Prince E, Sonuga-Barke E, Warner J, Stevenson J (2007) Food additives and hyperactive behaviour in 3-year-old and 8/9-year-old children in the community: a randomised, double-blinded, placebo-controlled trial. Lancet 370:1560–1567
McClain C, Cohen D (1989) Increased tumor necrosis factor production by monocytes in alcoholic hepatitis. Hepatology 9:349–351
McClain C, Hill D, Song Z, Chawla R, Watson W, Chen T, Barve S (2002) S-adenosylmethionine, cytokines, and alcoholic liver disease. Alcohol 27:185–192
Melis M, Camarini R, Ungless M, Bonci A (2002) Long-lasting potentiation of GABAergic synapses in dopamine neurons after a single in vivo ethanol exposure. J Neurosci 22:2074–2082
Mick E, Biederman J, Faraone S, Sayer J, Kleinman S (2002) Case-control study of attention-deficit hyperactivity disorder and maternal smoking, alcohol use, and drug use during pregnancy. J Am Acad Child Adolesc Psychiatry 41:378–385
Mill J, Richards S, Knight J, Curran S, Taylor E, Asherson P (2004) Haplotype analysis of SNAP-25 suggests a role in the aetiology of ADHD. Mol Psychiatry 9:801–810
Miller VM, Lewis DA, Rud KS, Offord KP, Croghan IT, Hurt RD (1998) Plasma nitric oxide before and after smoking cessation with nicotine nasal spray. J Clin Pharmacol 38:22–27
Min S, Moon I, Ko R, Shin H (2001) Effects of transdermal nicotine on attention and memory in healthy elderly non-smokers. Psychopharmacology (Berl) 159:83–88
Misener V, Schachar R, Ickowicz A, Malone M, Roberts W, Tannock R, Kennedy J, Pathare T, Barr C (2004) Replication test for association of the IL-1 receptor antagonist gene, IL1RN, with attention-deficit/hyperactivity disorder. Neuropsychobiology 50:231–234
Monje M, Toda H, Palmer T (2003) Inflammatory blockade restores adult hippocampal neurogenesis. Science 302:1760–1765
Morbini P, Villa C, Campo I, Zorzetto M, Inghilleri S, Luisetti M (2006) The receptor for advanced glycation end products and its ligands: a new inflammatory pathway in lung disease? Mod Pathol 19:1437–1445
Nabata A, Kuroki M, Ueba H, Hashimoto S, Umemoto T, Wada H, Yasu T, Saito M, Momomura S, Kawakami M (2008) C-reactive protein induces endothelial cell apoptosis and matrix metalloproteinase-9 production in human mononuclear cells: implications for the destabilization of atherosclerotic plaque. Atherosclerosis 196:129–135
Node K, Kitakaze M, Yoshikawa H, Kosaka H, Hori M (1997) Reversible reduction in plasma concentration of nitric oxide induced by cigarette smoking in young adults. Am J Cardiol 79:1538–1541
Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG (2000) Multiple sclerosis. N Engl J Med 343:938–952
Osaka H, Mukherjee P, Aisen P, Pasinetti G (1999) Complement-derived anaphylatoxin C5a protects against glutamate-mediated neurotoxicity. J Cell Biochem 73:303–311
Paine T, Neve R, Carlezon WJ (2009) Attention deficits and hyperactivity following inhibition of cAMP-dependent protein kinase within the medial prefrontal cortex of rats. Neuropsychopharmacology 34:2143–2155
Pasinetti G (1996) Inflammatory mechanisms in neurodegeneration and Alzheimer’s disease: the role of the complement system. Neurobiol Aging 17:707–716
Qin L, He J, Hanes R, Pluzarev O, Hong J, Crews F (2008) Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. J Neuroinflammation 5:10
Raz R, Gabis L (2009) Essential fatty acids and attention-deficit-hyperactivity disorder: a systematic review. Dev Med Child Neurol 51:580–592
Redford EJ, Kapoor R, Smith KJ (1997) Nitric oxide donors reversibly block axonal conduction: demyelinated axons are especially susceptible. Brain 120(Pt 12):2149–2157
Reif A, Rösler M, Freitag C, Schneider M, Eujen A, Kissling C, Wenzler D, Jacob C, Retz-Junginger P, Thome J, Lesch K, Retz W (2007) Nature and nurture predispose to violent behavior: serotonergic genes and adverse childhood environment. Neuropsychopharmacology 32:2375–2383
Reif A, Jacob C, Rujescu D, Herterich S, Lang S, Gutknecht L, Baehne C, Strobel A, Freitag C, Giegling I, Romanos M, Hartmann A, Rösler M, Renner T, Fallgatter A, Retz W, Ehlis A, Lesch K (2009) Influence of functional variant of neuronal nitric oxide synthase on impulsive behaviors in humans. Arch Gen Psychiatry 66:41–50
Retz W, Reif A, Freitag C, Retz-Junginger P, Rösler M (2010) Association of a functional variant of neuronal nitric oxide synthase gene with self-reported impulsiveness, venturesomeness and empathy in male offenders. J Neural Transm 117:321–324
Rezaee F, Rellick S, Piedimonte G, Akers S, O’Leary H, Martin K, Craig M, Gibson L (2010) Neurotrophins regulate bone marrow stromal cell IL-6 expression through the MAPK pathway. PLoS One 5:e9690
Ribasés M, Hervás A, Ramos-Quiroga J, Bosch R, Bielsa A, Gastaminza X, Fernández-Anguiano M, Nogueira M, Gómez-Barros N, Valero S, Gratacòs M, Estivill X, Casas M, Cormand B, Bayés M (2008) Association study of 10 genes encoding neurotrophic factors and their receptors in adult and child attention-deficit/hyperactivity disorder. Biol Psychiatry 63:935–945
Riediger N, Othman R, Suh M, Moghadasian M (2009) A systemic review of the roles of n-3 fatty acids in health and disease. J Am Diet Assoc 109:668–679
Romanos M, Gerlach M, Warnke A, Schmitt J (2010) Association of attention-deficit/hyperactivity disorder and atopic eczema modified by sleep disturbance in a large population-based sample. J Epidemiol Community Health 64:269–273
Rushforth S, Allison C, Wonnacott S, Shoaib M (2010) Subtype-selective nicotinic agonists enhance olfactory working memory in normal rats: a novel use of the odour span task. Neurosci Lett 471:114–118
Sarkar R, Gelabert HA, Mohiuddin KR, Thakor DK, Santibanez-Gallerani AS (1999) Effect of cigarette smoke on endothelial regeneration in vivo and nitric oxide levels. J Surg Res 82:43–47
Schlaepfer I, Clegg H, Corley R, Crowley T, Hewitt J, Hopfer C, Krauter K, Lessem J, Rhee S, Stallings M, Wehner J, Young S, Ehringer M (2007) The human protein kinase C gamma gene (PRKCG) as a susceptibility locus for behavioral disinhibition. Addict Biol 12:200–209
Schmitt J, Romanos M, Schmitt N, Meurer M, Kirch W (2009) Atopic eczema and attention-deficit/hyperactivity disorder in a population-based sample of children and adolescents. JAMA 301:724–726
Schmitt J, Apfelbacher C, Chen C, Romanos M, Sausenthaler S, Koletzko S, Bauer C, Hoffmann U, Krämer U, Berdel D, von Berg A, Wichmann H, Heinrich J, Group GINIpS (2010) Infant-onset eczema in relation to mental health problems at age 10 years: results from a prospective birth cohort study (German Infant Nutrition Intervention plus). J Allergy Clin Immunol 125:404–410
Segman R, Meltzer A, Gross-Tsur V, Kosov A, Frisch A, Inbar E, Darvasi A, Levy S, Goltser T, Weizman A, Galili-Weisstub E (2002) Preferential transmission of interleukin-1 receptor antagonist alleles in attention deficit hyperactivity disorder. Mol Psychiatry 7:72–74
Sen J, Belli A (2007) S100B in neuropathologic states: the CRP of the brain? J Neurosci Res 85:1373–1380
Sharp S, McQuillin A, Gurling H (2009) Genetics of attention-deficit hyperactivity disorder (ADHD). Neuropharmacology 57:590–600
Shinawi M, Liu P, Kang S, Shen J, Belmont J, Scott D, Probst F, Craigen W, Graham B, Pursley A, Clark G, Lee J, Proud M, Stocco A, Rodriguez D, Kozel B, Sparagana S, Roeder E, McGrew S, Kurczynski T, Allison L, Amato S, Savage S, Patel A, Stankiewicz P, Beaudet A, Cheung S, Lupski J (2009) Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioral problems, dysmorphism, epilepsy, and abnormal head size. J Med Genet 47:332–341
Shiow L, Paris K, Akana M, Cyster J, Sorensen R, Puck J (2009) Severe combined immunodeficiency (SCID) and attention deficit hyperactivity disorder (ADHD) associated with a Coronin-1A mutation and a chromosome 16p11.2 deletion. Clin Immunol 131:24–30
Shohami E, Novikov M, Bass R, Yamin A, Gallily R (1994) Closed head injury triggers early production of TNF alpha and IL-6 by brain tissue. J Cereb Blood Flow Metab 14:615–619
Silva C, Rio M, Cruz C (1985) Local immunoglobulin synthesis and blood-brain barrier assessment in subacute sclerosing panencephalitis. Eur Neurol 24:128–133
Smith KJ, Kapoor R, Hall SM, Davies M (2001) Electrically active axons degenerate when exposed to nitric oxide. Ann Neurol 49:470–476
Smith M, Moylan J, Smith J, Li W, Reid M (2007) IFN-gamma does not mimic the catabolic effects of TNF-alpha. Am J Physiol Cell Physiol 293:C1947–C1952
Spector R (1988) Fatty-acid transport through the blood-brain barrier. J Neurochem 50:639–643
Stillwell W, Shaikh S, Zerouga M, Siddiqui R, Wassall S (2005) Docosahexaenoic acid affects cell signaling by altering lipid rafts. Reprod Nutr Dev 45:559–579
Tai C, Chen C, Lee H, Wang Y, Li T, Mersamm H, Ding S, Wang P (2009) Docosahexaenoic acid enhances hepatic serum amyloid A expression via protein kinase A-dependent mechanism. J Biol Chem 284:32239–32247
Taupin V, Toulmond S, Serrano A, Benavides J, Zavala F (1993) Increase in IL-6, IL-1 and TNF levels in rat brain following traumatic lesion. Influence of pre- and post-traumatic treatment with Ro5 4864, a peripheral-type (p site) benzodiazepine ligand. J Neuroimmunol 42:177–185
Thapar A, Rice F, Hay D, Boivin J, Langley K, van den Bree M, Rutter M, Harold G (2009) Prenatal smoking might not cause attention-deficit/hyperactivity disorder: evidence from a novel design. Biol Psychiatry 66:722–727
Tikka T, Fiebich B, Goldsteins G, Keinanen R, Koistinaho J (2001) Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci 21:2580–2588
Toga A, Thompson P, Sowell E (2006) Mapping brain maturation. Trends Neurosci 29:148–159
Vincent S (2010) Nitric oxide neurons and neurotransmission. Prog Neurobiol 90:246–255
Wan J, Fu A, Ip F, Ng H, Hugon J, Page G, Wang J, Lai K, Wu Z, Ip N (2010) Tyk2/STAT3 signaling mediates beta-amyloid-induced neuronal cell death: implications in Alzheimer’s disease. J Neurosci 30:6873–6881
Wang H, Zakhari S, Jung M (2010) Alcohol, inflammation, and gut-liver-brain interactions in tissue damage and disease development. World J Gastroenterol 16:1304–1313
Yan S, Ramasamy R, Naka Y, Schmidt A (2003) Glycation, inflammation, and RAGE: a scaffold for the macrovascular complications of diabetes and beyond. Circ Res 93:1159–1169
Yan T, McQuillin A, Thapar A, Asherson P, Hunt S, Stanford S, Gurling H (2010) NK1 (TACR1) receptor gene ‘knockout’ mouse phenotype predicts genetic association with ADHD. J Psychopharmacol 24:27–38
Zhou JF, Yan XF, Guo FZ, Sun NY, Qian ZJ, Ding DY (2000) Effects of cigarette smoking and smoking cessation on plasma constituents and enzyme activities related to oxidative stress. Biomed Environ Sci 13:44–55
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Donev, R., Thome, J. Inflammation: good or bad for ADHD?. ADHD Atten Def Hyp Disord 2, 257–266 (2010). https://doi.org/10.1007/s12402-010-0038-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12402-010-0038-7