
Abstract
The present study discussed the application of the intercropping system to improve land use efficacy and soil microbial activity. We assessed linkages of soil properties and unculturable diazotrophs community under three cultivation systems (monoculture sugarcane, peanut–sugarcane and soybean–sugarcane intercropping). Rhizosphere soil of sugarcane was sampled and DNA was extracted. We amplified the nifH gene and sequenced by high throughput sequencing. The bioinformatics analysis of sequenced data obtained a total of 436,458 nifH gene reads that are classified into 3201 unique operational taxonomic units (OTUs). A higher percentage of exclusive OTUs identified under soybean–sugarcane intercropping (< 375). The microbial structure results showed that Alpha-proteobacteria and Beta-proteobacteria were the dominant groups in all three cultivation systems. While genus such as Bradyrhizobium, Burkholderia, Pelomonas, and Sphingomonas was predominant in the intercropping systems and these diazotrophic bacterial communities were positively correlated to the soil pH and soil enzyme protease. Additionally, a lower quantity of available P in the soil of intercrops indicated a strong link between soil nutrients uptake and microbial activity. The results of the present study concluded some interesting facts of intercropping systems that positively improved the soil microbial activity and this kind of strategy could help to cultivate multiple crops to improve the economic growth of the country by sustainable sugarcane production.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Sugarcane (Saccharum species hybrid) known as sugar crop that nurtured globally in the tropical and subtropical areas (Li et al. 2015). Sugarcane requires substantial amounts of nitrogen-containing fertilizers for optimum growth and crop production (Yang et al. 2013). However, in recent year’s sugarcane growers’ facing several problems due to overuse of N fertilizer such as higher production cost, soil infertility, and it also raised environmental pollution. Moreover, it can be dangerous to the environment through the emission of N-containing gases and soluble N compounds polluted the groundwater, which can lead to the eutrophication (Robertson and Vitousek 2009). Prokaryotic microorganisms that have the ability to fix nitrogen known as diazotrophs, these microbes played a significant role to convert atmospheric nitrogen gas as biologically available ammonium and it provides numerous advantages for the plants (Solanki et al. 2018). Several leguminous plants like soybean, peanut, pea, chickpea, etc. are well known for N fixation and recently, rhizospheric and endophytic diazotrophic bacteria have been also isolated from various nonleguminous plants, including rice (Xie and Yokota 2006), maize (Perin et al. 2006), and sugarcane (Solanki et al. 2018). Among different soil management practices, intercropping has also played a significant role in soil mineralization and utilized all natural resources far better than a single crop.
Yang et al. (2013) reported that sugarcane–legume intercrops can utilize soil and atmospheric N efficiently and improve the production and quality with less fertilizer application that also protects the environment, and several reports showed the intercrops caused significant changes in soil properties and microbial population (Wang et al. 2014; Solanki et al. 2018). Sugarcane plants are also fixing the atmospheric nitrogen through N2-fixation allied with diazotrophic bacteria (Perin et al. 2006; Solanki et al. 2018). Maximum diazotrophs that accomplish biological nitrogen fixation (BNF) with the nitrogen fixation gene (nifH) cluster, which is a highly conserved functional gene encodes the iron protein of nitrogenase (Zehr et al. 2003) and has been thoroughly studied from environmental DNA samples (Coelho et al. 2009). It is a convenient marker in culture-independent studies, to provide evidence for significant N fixation in diverse environments (Coelho et al. 2009; Zou et al. 2011). To date, culture-independent methods have investigated N fixation in different habitats, including soils (Li et al. 2012), plant parts (Solanki et al. 2018) and water resources (Blais et al. 2012; Tai et al. 2013). By focusing on the sequences of the nifH genes, large numbers of sequence data have been also obtained by independent culture methods (Gaby and Buckley 2014). Since it is claimed that some of these databases are also suitable for molecular analysis, it is imaginable that they might be used to study the diazotrophic community by means of Next-generation sequencing. A diazotrophic bacterial community can be affected by numerous factors. Moreover, plant biomass, the chemical composition of the litter and root exudates may be playing the role as an intermediate for the composition of diazotrophic bacteria. Soil physicochemical factors, like pH and water holding capacity, microbial biomass carbon and nitrogen, total nitrogen, potassium, phosphorus and available phosphorus, electrical conductivity and soil nutrient availability, have also been identified as drivers of nifH gene diversity and abundance in different environments (Blais et al. 2012; Li et al. 2012; Tai et al. 2013).
However, to the best of our knowledge, no study has been explored in the sugarcane–legume intercropping rhizospheric soil to characterize the diazotrophic community by using high throughput sequencing. Therefore, the economic importance of diazotrophic microbes under intercropping systems remains unexplored. Microbial mapping and importance of diazotrophic microbes would provide essential information to our understanding about the significance of diazotrophs in rhizospheric N pool. In our previous study, we already determined that intercropping system improved the diazotrophic population in soil by identification of cultivable microbes (Solanki et al. 2017). Therefore, in the present study, we aimed to (1) explore the unculturable diazotrophic bacteria in the intercropping system via next-generation sequencing and (2) their relationship with the soil properties.
Materials and Methods
Soil Sampling
Sugarcane rhizosphere soil sampling was carried out in April 2014, during the vegetative stage at the farmer fields and research stations in the Guangxi Province, China (Table S1). Soil samples were collected up to 30 cm depth using an auger (3.5 cm in diameter) in close proximity of the roots of sugarcane monoculture (M) fields as well as sugarcane + peanut (SP) and sugarcane + soybean (SS) intercropped fields. Three different plots of monoculture and intercropping were evaluated at each location and five plants were randomly selected and sampled from each plot, then mixed together to give a composite soil sample. A total of 3 samples from each location sampled. All soil samples were stored in the icebox and sent immediately to the lab within the following hours. Air-dried composite sample was sieved through a 2.0 mm mesh and plant and roots residues were removed by hand earlier to chemical analysis.
Soil Analysis
Soil chemical properties and soil enzymes (urease, protease, and nitrite reductase) were analyzed in the soil analysis laboratory of Guangxi University, Nanning, Guangxi Province, China. Soil pH analysis (soil: water = 1:1) performed by pH meter and soil organic carbon was assessed by dichromate oxidation (Walkley and Black 1934). Total N was detected through Semimicro-Kjeldahl method (Bremmer and Mulvaney 1982). Nitrate nitrogen (NO3−–N) and exchangeable ammonium nitrogen (NH4+–N) was estimated by FeSO4/Zn reduction method of Carter (Carter 1993). Total phosphorus (P) was measured via Sodium carbonate fusion defined by Carter (Carter 1993). Available P was estimated through samples extraction method 0.5 M NaHCO3, and determining P calorimetrically via molybdate (Bao 2002). Total K was estimated using a Flame photometry method (Bao 2002). Available K was estimated using 1 N ammonium acetate extraction-flame photometry method (Bao 2002). Urease activity was assessed via phenol-sodium hypochlorite calorimetry and expressed as mg NH3–N Kg−1 d−1 (Guan et al. 1986). Nitrite reductase activity was examined using the Gerry reagent method and expressed as mg NO2–N g−1 d−1 (Li et al. 2008). Protease activity was determined and expressed as mg Kg−1 d−1 (Ruan et al. 2009). Other soil enzymes like dehydrogenase (DHA) and nitrogenase were assessed by the following methods. Soil dehydrogenase activity was assessed by the method of Singh and Singh, 2005. Nitrogen fixation activity was measured using the acetylene reduction assay (ARA) (Hardy et al. 1968). All soil samples were incubated individually with 10% acetylene for 3 h. Autoclaved distilled water incubated with 10% acetylene used as a control. The measurements were performed via a gas chromatograph (GC-17A, Shimadzu, Kyoto, Japan) furnished with a flame ionization detector and a column filled with DB-1701 (Agilent, Santa Clara, USA).
DNA Extraction, Gene Amplification, and Illumina-Sequencing
Genomic DNA was extracted from three independent soil samples and pooled together. After pooling, purification was performed using the Ezup Column Soil DNA Purification Kit (Sangon Biotech, Shanghai, China). DNA quality and concentration were evaluated using a NanoDrop ND-2000 UV–Vis Spectrophotometer (Thermo Scientific, Wilmington, DE, USA), based on the ratios of 260 nm/280 nm and 260 nm/230 nm. Amplification of partial nifH sequences was performed with the degenerate Z-primers (Zehr et al. 1998) by the Nested polymerase chain reaction. Ready-to-use 2 × concentrated PCR masters (0.1 U μL−1 TaqDNA polymerase, 0.2 mM dNTP, 3 mM MgCl2, 2 × PCR buffer) obtained by Tiangen (Tiangen Biotech, Beijing, China) was used for PCR reaction. PCR amplification was performed with a Peltier Thermal Cycler (Bio-Rad, Hercules, USA). First step PCR was performed with outer primer (nifH3 and nifH4). The amplification protocol consisted of an initial denaturing step at 94 °C for 4 min, then 30 cycles of 1 min each at 94 °C, 55 °C, and 72 °C, followed by a final extension at 72 °C for 7 min. The second step PCR entailed the same concentration of reagents as the initial PCR reaction, using first PCR-product as a template with inner primers (nifH1 and nifH2). A touchdown PCR strategy was used for nifH amplification to improve amplification specificity. In the first 20 touchdown cycles, the annealing temperature declined by 0.5 °C per cycle from 67 to 57 °C. Further fifteen cycles were performed at the annealing temperature of 57 °C. PCR products were electrophoresed on agarose gels, stained with ethidium bromide, visualized to check the quality. The expected amplicons were excised from agarose gels, purified by TIANgel Midi Purification Kit (Tiangen Biotech, Beijing, China). The amplicons were used to construct libraries after qualified. The jagged ends of the fragment would be changed into blunt ends by using T4 DNA polymerase, Klenow Fragment, and T4 Polynucleotide Kinase. Then an ‘A’ base was added to each 3’ end to make it easier to add adapters. After all that, fragments too short would be removed by AMPure beads, only the qualified library can be used for sequencing by Genome Analyzer/HiSeq (Illumina-Solexa, San Diego, USA).
Bioinformatics
To enhance the accuracy and reliability of results through Bioinformatics analysis, the raw sequence data were processed by the method described by Fadrosh et al. 2014. The high-quality paired-end reads were combined to tags based on overlaps by FLASH software (Magoc and Salzberg 2011). The tags were clustered to OTUs (Operational Taxonomic Unit) by scripts of software USEARCH with a 97% similarity threshold (Edgar 2013), Chimeras were filtered out by using UCHIME (Edgar et al. 2011), then the OTU unique representative sequences were obtained. All tags were mapped to each OTU representative sequences using USEARCH GLOBAL (Wang et al. 2007), and then the tags’ number of each OTU in each sample will be summarized to OTU abundance table. Taxonomic ranks were allocated to OTU representative sequence using the Ribosomal Database Project (RDP) (Cole et al. 2014) trained on ARB nifH database (Heller et al. 2014). At last, alpha diversity, beta diversity and the species distribution were analyzed based on OTU and taxonomic ranks. Meta-sequence data were deposited with the NCBI under Bio-Project accession Number PRJNA310619.
Statistical Analysis
Box plot of soil properties performed by using Origin 2017SR2 (Northampton, MA, USA). Based on the OTU abundance, Venn diagram, principal component analysis (PCA) (package-ade4) and rank curve were drawn by software R (v3.0.3). The PCA was used to find variability and correlation among the OUT abundance of different cultivation systems. The indices are calculated through Mothur (v1.31.2), and the corresponding rarefaction curves were drawn by Origin 2017SR2. A beta diversity heat map was drawn by ‘aheatmap’ in package ‘NMF’ of software R (v3.0.3). The tags number of each taxonomic rank (Phylum, Class, Order, Family, Genus, and Species) or OTU in different samples were summarized with the software R (v3.0.3). Species heat map analysis was done based on the relative abundance of each species in each sample. To minimize the degree of the difference of the relative abundance value, the values were all log transformed. If the relative abundance of certain species is 0, the half of the minimum abundance value will substitute for it. Heat maps were generated using the package ‘gplots’ of software R (v3.0.3) and the distance algorithm is ‘euclidean’, the clustering method is ‘complete’. A representative OTU phylogenetic tree was constructed using the QIIME (v1.80) built-in scripts, including the fast tree method for tree construction and phylogeny tree was imaged by software R (v3.0.3) at last. Correlation and Canonical correspondence analysis (CCA) was performed to link microbial community and soil variables by using Past3 software (Hammer et al. 2001). CCA is a multivariate constrained ordination technique that extracts major gradients among combinations of explanatory variables in a dataset. CCA can help to extract overall assemblage structure and relationships between community structure and soil variables.
Results
Soil Parameters
Box plots for the soil pH, organic matter (OM), soil nutrient (Total N, P, K, N (NO3), N (NH4), available P and K) and enzymes (nitrogenase, nitrite reductase, urease, protease and dehydrogenase) contents of the samples representing the tree cropping system were displayed in Fig. 1. Lower pH was found in monoculture than both intercropping. Higher OM content was found in peanut intercropping than monoculture and soybean intercropping. Similar, Intercropping system showed a higher average of total N, total K, N (NH4), available K, nitrite reductase, protease, dehydrogenase, and nitrogenase (Fig. 1). The differences in soil chemical properties between cropping systems encouraged us to carry out separate analyses for diazotrophic microbial communities.

The distribution of soil chemical and biological properties among the three cultivation systems. M-sugarcane monoculture, P-peanut–sugarcane intercropping and S-soybean–sugarcane intercropping
Distribution, Diversity, and Richness of Operational Taxonomic Units
After sequencing, 522,223 raw sequenced reads were obtained and after trimming their 436,458 effective reads were recovered from all soil samples with an average 83.66% read utilization ratio (Table S2). The high-quality paired-end reads were combined to tags based on overlaps, 435,439 tags were obtained in total with 39,585 tags per sample on average, and the average length is 358–366 bp with approx. 99% connecting ratio. Chimera filtered tags were assembled into Operational Taxonomic Units (OTU) at 97% similarity and a total of 3201 OTUs was acquired. Outs rank curved showed maximum OTUs recovered with MISS and less with MIM (Figure S1). Cultivation systems wise maximum OTUs resulted in soybean–sugarcane intercropping (< 376) and followed by monoculture (< 263) and peanut–sugarcane intercropping showed less OTUs (< 254) (Table S3). In order to display the differences of OTU composition of different rhizospheric samples, PCA was used and a scattered PCA plot showed a similarity between soil samples of all locations except MIM and minor difference on the basis of cultivation system except MIM (Figure S1). In order to find out the apparent uniformity recommended by the PCA, we calculated alpha diversity to recognize the differences among bacterial communities from different cultivation systems. In soybean–sugarcane intercropping, the analyses of different richness and diversity indices revealed a slight increase in richness in communities from monoculture, and lower community diversity in peanut–sugarcane intercropping (Tables S3) and cultivation systems based box plot on observed species and Shannon’s index also showed the similar results (Fig. 2a, b). Similarly, the rarefaction curves showed richness with soybean intercropping (Figure S2). To conclude the similarities and differences in community structure among three cultivation systems, pairwise Bray–Curtis dissimilarities were calculated and ordinated in scattered plot and diversity heat map (Fig. 3a). The PCA plot of Bray–Curtis dissimilarity showed higher diversity among cultivations at different locations MI and MY. However, W and CT showed less diversity (Fig. 3a).

Alpha diversity box-plot among three cultivation systems. a Observed species and b Shannon’s Index, M—sugarcane monoculture, P—peanut–sugarcane intercropping and S—soybean–sugarcane intercropping

a Beta diversity metrics among three cultivation systems, b Venn diagrams showing the overall overlap of OTUs between three cultivation systems (M, P, and S) obtained in four different locations. M—sugarcane monoculture, P—peanut–sugarcane intercropping and S—soybean–sugarcane intercropping
Moreover, based on the OTU abundance of different cultivation systems, OTU of each group was listed and a Venn diagram was drawn. Venn diagram showed maximum unique OTUs were found in soybean–sugarcane intercropping and followed by monoculture and peanut–sugarcane intercropping. Maximum overlapped OTUs (284) identified among soybean–sugarcane intercropping and monoculture and followed by monoculture and peanut–sugarcane intercropping and 266 OTUs were overlapped in all three cultivation systems (Fig. 3b).
Diversity of Bacterial Communities
A total of 436,458 high-quality sequences of nifH gene with an average length of 360 bp were obtained from the all samples, and all sequences belonging to Proteobacteria phyla, two classes (Alpha-proteobacteria and Beta-proteobacteria), three orders (Burkholderiales, Rhizobiales and Sphingomonadales), seven families (Alcaligenaceae, Bradyrhizobiaceae, Burkholderiaceae, Comamonadaceae, Methylocystaceae, Rhizobiaceae and Sphingomonadaceae), seven genera (Azohydromonas, Bradyrhizobium, Burkholderia, Pelomonas, Pseudacidovorax, Rhizobium/Agrobacterium group and Sphingomonas), and five species (Burkholderia silvatlantica, Pelomonas saccharophila, Pseudacidovorax sp. ptl-2, Rhizobium sp. W3 and Sphingomonas azotifigens) were identified among all cultivation systems of four locations, and a major percentage of sequence data were unclassified. Next-generation sequencing results have clearly distinguished the differences among cultivation systems on the basis of their bacterial community compositions. Changes in rhizospheric diazotrophic bacterial community compositions were also noticed among three cultivations, showing some variation of the abundance of each group by the locations. Maximum average abundance of Proteobacteria was identified with peanut–sugarcane intercropping and followed by soybean–sugarcane intercropping and monoculture. However, the maximum abundance of Alpha-proteobacteria was recovered with monoculture and Beta-proteobacteria with peanut–sugarcane intercropping. On the basis of order, a maximum abundance of Burkholderiales was resulted in peanut–sugarcane intercropping, Rhizobiales with monoculture and Sphingomonadales with soybean–sugarcane intercropping, respectively. Different patterns of community structure were observed at the family and genera levels across all sites (Fig. 4a, b). Among all the samples, the highest abundance was identified in family Rhizobiaceae and followed by Alcaligenaceae, Burkholderiaceae, Comamonadaceae, Bradyrhizobiaceae, Sphingomonadaceae, and Methylocystaceae, respectively. Maximum abundance of family Alcaligenaceae, Bradyrhizobiaceae and Comamonadaceae were resulted with peanut–sugarcane intercropping, and family Burkholderiaceae and Sphingomonadaceae were identified with soybean–sugarcane intercropping. However, family Rhizobiaceae and Methylocystaceae resulted in monoculture (Fig. 4). Similar to the family, higher abundance resulted with the genus Rhizobium/Agrobacterium group and followed by Azohydromonas, Burkholderia, Bradyrhizobium, Pelomonas, Sphingomona, and Pseudacidovorax, respectively (Fig. 4b).

Relative distribution of diazotrophic bacterial taxa at the family level (a) and genera level (b) among three cultivation systems samples from four different locations of Guangxi, China, M—sugarcane monoculture, P—peanut–sugarcane intercropping and S—soybean–sugarcane intercropping
Moreover, heat maps were determined to get a graphical colored matrix of individual sample sequence data. Heat map displayed abundance based genus/species clustering of each genus/species, longitudinal clustering specifies the similarity of all genus/species between the samples, and the horizontal clustering directs the similarity of certain genus/species among the samples, the closer the distance and the shorter the branch length is, the more similar the genus/species composition between the samples (Fig. 5). Heat map showed clusters in both intercropping cultivations and positive influence of three genus Azohydromonas, Pseudacidovorax, and Sphingomonas determined in soybean intercropping, and Bradyrhizobium, and Pelomonas determined in peanut intercropping (Fig. 5a). Next to this, species abundance based heat map also showed two major clusters among species and different cultivation systems. Pelomonas_saccharophila, Pseudacidovorax_sp._ptl-2, and Sphingomonas_azotifigens showed higher abundance via dark spots in both intercropping cultivations (Fig. 5b).

Relative abundance heatmap of diazotrophic bacterial taxa at genes (a) and species (b) levels among three cultivation samples from four different locations of Guangxi, China. Longitudinal clustering indicates the similarity of all species among cropping systems, and the horizontal clustering indicates the similarity of certain species among different cropping systems. The closer the distance is and the shorter the branch length is, the more similar the species composition is between the samples. M—sugarcane monoculture, P—peanut–sugarcane intercropping and S—soybean–sugarcane intercropping
Linking Between Soil Properties and Microbial Community
Soil pH positively related to protease. Total N–P–K, N–NO3, available K, and nitrogenease were positively related to soil organic matter (Table 1; Fig. 6). On the contrary, soil organic matter showed a significant negative correlation with nitrite reductase. Among soil minerals, total N showed a significant positive correlation with total P and available K, and total K with N–NO3 and N–NH4, whereas a strong negative correlation was found with total P and nitrite reductase. Moreover, nitrogenase positively correlated with total N, total K, and N–NO3. However, urease showed a strong negative correlation with N–NH4 (Table 1).

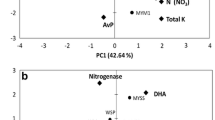
Ordination diagrams from canonical correspondence analysis (CCA) of microbial abundances and soil parameters. Blue and green arrows indicate the direction and magnitude of soil chemical and biological parameters associated with microbial abundance. Sugarcane monoculture (M—diamond), peanut–sugarcane intercropping (P—triangle) and soybean–sugarcane intercropping (S—square), red dot for the taxa. Azo Azohydromonas, Brad Bradyrhizobium, Bur Burkholderia, Pel Pelomonas, Ps Pseudacidovorax, Rhiz Rhizobium/Agrobacterium group and Sp Sphingomonas, OM organic matter, Av available, Ur Urease, Nr Nitrite reductase, Pr Protease, Dh Dehydrogenase, and Ng Nitrogenase
Soil pH positively related to Azohydromonas and OM positively related with Bradyrhizobium, whereas soil pH negative correlation was found with Burkholderia (Table 2). For soil minerals, total N–P showed a significant positive correlation with Bradyrhizobium and total K showed a positive correlation with Sphingomonas. N–NH4 showed a positive correlation with Pelomonas and a negative correlation found with the Shannon diversity index, whereas available P positively correlated with Rhizobium/Agrobacterium and available K showed a positive correlation with Bradyrhizobium and Sphingomonas. Among soil enzymes, protease positively correlated with Azohydromonas. On the contrary, urease, nitrite reductase and protease a negatively correlated with Pelomonas, Bradyrhizobium, Burkholderia and Rhizobium/Agrobacterium, respectively (Table 2).
To further discover these relations, we performed a canonical correlation analysis (CCA) based on the microbial abundance and soil variables (Fig. 6). CCA ordination showed that soil properties positively influenced the microbial composition. The results of the CCA showed a positive correlation of soil pH, available K, total N, N–NO3 and protease with CCA axis 1, and based on magnitude and position they were important for explaining the variations between the microbial communities. The relatively small magnitude of the OM, total P, total K, N–NH4, nitrite reductase, nitrogenase, urease, and dehydrogenase vectors indicates that these parameters were not as strongly correlated to community composition than other (higher magnitude vectors). However, a higher magnitude of available P showed a negative correlation as compared to other soil variables. Moreover, some bacterial genera, Azohydromonas, Bradyrhizobium, Burkholderia, Pseudacidovorax, Rhizobium and Sphingomonas positively correlated with soil chemical and biological properties. Azohydromonas and Rhizobium were most distinguished genus resulted by CCA. On the contrary, Pelomonas showed a negative correlation with all soil properties (Fig. 6).
Discussion
To understand the functional diversity, composition, structure, and dynamics of diazotrophs communities under sugarcane–legume intercropping. We carried out this study to examine the effect of different cultivation systems on the soil properties and their relation to the diazotrophs. Previous studies have investigated that soil microbial diversity, soil enzyme activities, crop yield could be affected by land management practices, and all these soil properties were increased under intercropping systems than monoculture (Zou et al. 2011; Yang et al. 2013; Wang et al. 2014). Moreover, our previous study also discussed that soil biochemical and enzymes have a direct effect by an intercropping system and it enhanced the diazotrophic bacterial population (Solanki et al. 2017; 2018). Kent and Triplett (2002) have specified that root secretions and rhizodeposition of different plant root zones influenced the structural and functional diversity of rhizosphere. Furthermore, the soil type, cropping practices, plant growth stages, and ecological factors influence the rhizospheric microbial community (Li et al. 2012). Our results also correlated with these studies. Soil enzymes are essential for nutrient cycling. Soil enzyme activity is also an important bioindicator of soil fertility and quality. Activities of soil urease, nitrite reductase, protease, dehydrogenase, and nitrogenase are important indexes for evaluating biochemical processes related to soil N transformation. In our study, monoculture showed higher urease and intercropping samples were showed higher activity of nitrite reductase, protease, dehydrogenase, and nitrogenase (Fig. 1). One possibility is that in monoculture for higher urease activity, N-fertilizer rate was the same in monoculture and intercrops, and intercrops utilized soil urea faster than monoculture. Nitrite reductase, protease, and nitrogenase play significant roles in the nitrogen cycle and higher activities of these enzymes showed that vital microbial community enhanced in intercropping system. Reduction of atmospheric dinitrogen into the ammonia by nitrogenase enzyme is an energy consuming process, therefore, it’s not easily regulated and favorable environmental factors like acidic pH, less available N concentration and rich C sources will increase free-living N-fixation (Orr et al. 2011). The positive correlation of pH with microbial population showed similarity with the past studies (Table 1; Fig. 6) (Orr et al. 2011; Zou et al. 2011; Yousuf et al. 2014). Recently, sequence-based community structure analysis is progressively accepted to study rhizospheric bacterial communities (Gaby and Buckley 2014). Through cultivation-independent molecular technique, we deliver an evaluation of reaction and recovery of rhizospheric diazotrophs communities influenced by different cultivation systems. Moreover, in the present study nested PCR was used to amplify the functional nifH gene from the rhizospheric soil samples. Nested PCR decreases the chance of multiple target site amplifications and enhances detection consistency. The effectiveness of the nested PCR method has been verified several times in the previous nifH gene diversity studies (Orr et al. 2011; Blais et al. 2012; Li et al. 2012). Li et al. (2012) reported that terrestrial ecosystems had less abundance of diazotrophic bacteria therefore, direct amplification of the nifH gene from soil samples is hard. In the present study, we have been using nested PCR to enhance the sensitivity and the specificity of nifH gene amplification, to investigate the genetic diversity from rhizospheric soil samples. Our results showed that 436,458 high-quality sequences of nifH gene belonged to the Proteobacteria phyla. More specifically, these sequences were affiliated with the genera Rhizobium/Agrobacterium, Azohydromonas, Burkholderia, Bradyrhizobium, Pelomonas, Sphingomonas and Pseudacidovorax (Fig. 5a), all of which contain species that are known to fix nitrogen in symbioses with legumes (Sachs et al. 2009). The nifH gene sequences of the present study with the uncultivable diazotrophs of different rhizosphere, revealed common genera like Azohydromonas, Burkholderia, Bradyrhizobium and Rhizobium (Perin et al. 2006; Coelho et al. 2009; Yousuf et al. 2014), and our result also showed similarity with past studies by different molecular tools (Orr et al. 2011; Blais et al. 2012; Li et al. 2012). The high abundance of Alpha-proteobacteria (especially Burkholderia and Rhizobium) nifH genes found in the monoculture samples in farmer field, Mingyang Farm, Wuxu Town, District Jiangnan, Nanning City, due to the crop rotation and intercropping with leguminous plants (soybean and peanut) or watermelon. Moreover, Rhizobium sp. is well notorious root colonizer of a wide range of nonlegume plants like sugarcane (Solanki et al. 2018), sorghum (Coelho et al. 2009), rice (Xie and Yokota 2006) and maize (Perin et al. 2006), and Burkholderia sp. has been already detected as an active diazotrophs in sugarcane (Perin et al. 2006; Solanki et al. 2018). Regardless of diazotrophs, the most abundant taxa were Alpha-proteobacteria, and this outcome was usually consistent with past researchers who verified that Alpha-proteobacteria were the most abundant and common group in soil (Coelho et al. 2009; Li et al. 2014; Yousuf et al. 2014). Based on the observed nifH OTUs richness and calculated estimates, the sugarcane soil from the formers field is the most diverse in the soil sites included in this study. The predominance of nifH OTUs related to proteobacteria and the nonappearance of nifH gene sequence related to Cyanobacteria, Firmicutes, and Archaea has been also described from some previous studies (Coelho et al. 2009; Tai et al. 2013, 2014). Unexpectedly, there was no evidence of Gamma-proteobacterial nifH OTUs. The absence of these sequences is confirmed by the cultivation-dependent diazotrophs study from tropical soils where only Alpha-proteobacteria and Beta-proteobacteria have been found (Asis et al. 2000; Solanki et al. 2018). Next, we found Burkholderia silvatlantica, Pelomonas saccharophila, Pseudacidovorax sp., Rhizobium sp. W3 and Sphingomonas azotifigens species and they have well-known plant and soil associated microbes (Fig. 5b). Perin et al. (2006) reported a cultivable diazotrophs Burkholderia silvatlantica strain isolated from soil in Brazil. Pelomonas saccharophila strain also characterized as diazotrophs and hydrogen-oxidizing bacteria previously from Japan (Xie and Yokota 2005). Zhang and Chen (2012) isolated and characterized an N-fixers strain of Pseudacidovorax sp. from seawater of China. Kaur et al. (2011) reported an N-fixers Rhizobium sp. W3 strain isolated from hexachlorocyclohexane dumpsite of India. An N-fixer strain of Sphingomonas azotifigens was also isolated from rice plants in Japan (Xie and Yokota 2006). All these findings concluded that these species played important roles in the soil for the plant growth promotion and mineral acquisition.
The results obtained by other studies indicated that monoculture cultivation reduced the soil fertility and microbial diversity (negatively affected the structure, function, and stability of soil microflora), especially during the ratoon (Li and Yang 2015). Shannon’s diversity index of the nifH gene ranged 2.85–5.27 in this study (Fig. 2), which is higher than those in other studies (Tai et al. 2013, 2014). In the present study, soybean–sugarcane intercropping enhanced the values of species richness and diversity (Table S3). These results concluded that the monoculture cultivation system might alter the microenvironment of rhizospheric diazotrophs, and thus initiating soil bacterial community reductions. In our study, Pearson correlation analysis discovered the impact of soil properties on soil microbial diversity and community composition, which recommended that the variation in bacterial community abundance and diversity could be partly due to changes in the soil properties. Pearson correlation analysis and CCA plot revealed that the microbial community had positive links with the soil pH, nitrite reductase and protease. (Table 1; Fig. 6). Thus, the observed reductions in the soil pH and the activities of soil enzymes nitrite reductase and protease in monoculture could explain the declining in the diversity of microbial communities. Moreover, soil enzymes showed higher activity of nitrate reductase in intercropped soil samples and this enzyme played an essential role in denitrification. Denitrification is a process of nitrate reduction into a simple nitrogen molecule (nitric oxide) by diazotrophs and different bacterial species and enzymes have played an important role in this process (Song and Ward 2003). Likewise, intercropped fields showed higher protease activity, these results described the significant role of diazotrophic bacteria in N mineralization. Recently, Solanki et al. (2018) also testified the significant role of soil proteases in N mineralization, and this process played an important role for available N of plant and plant growth.
Microbial diversity analyses suggest that microbial composition is largely affected by soil parameters but provide little evidence about the linkage. Therefore, CCA was used to discover the link between microbial composition and soil variables. Soil total N, total P, OM, and N–NO3 appeared to be important soil variable and correlate positively. On the contrary, less available P in intercropping and negative correlation with microbial abundance showed that due to nitrogen-fixing microbes P uptake enhanced significantly and it reduced the soil available P. Our results agreed with the previous study of Li et al. (2016) reported that intercropping reduced the Olsen P in the soil. Moreover, higher phosphate uptake enhanced the plant growth and diazotrophic microbes like Bradyrhizobium, Burkholderia and Rhizobium are well-known phosphate solubilizer (Zeng et al. 2017).
In conclusion, this study presents a comparative study of the nifH gene biodiversity of sugarcane rhizosphere soil by next-generation sequencing in intercropped sugarcane. This study found that an extensive variety of diazotrophic bacteria were strongly collaborating with soil variable and intercropping cultivation system significantly enhanced the microbial diversity. Most of the soil variables positively correlated with diazotrophs. Maximum number of OTUs recovered from soybean–sugarcane intercropping and less available P of soil has given an idea to the utilization of intercropping system for nutrient mineralization, and we found that Alpha-proteobacteria abundantly recovered from the sugarcane rhizosphere. We concluded that soil physicochemical properties, especially nutrient elements, soil pH and soil enzymes positively influenced the bacterial community. However, these all result need to investigate further in regards to identify the important microbial candidates and need to understand their interaction with the plant to find out the nature of mechanisms of N cycling and P mineralization.
References
Asis Jr., C.A., M. Kubota, V.K. Chebotar, H. Ohta, Y. Arima, K. Nishiyama, K.I. Tsuchiya, and S. Akao. 2000. Endophytic bacterial population in Philippine sugarcane cultivars and isolation of nitrogen-fixing strains. Microbes and Environments, 15: 209–216. https://doi.org/10.1264/jsme2.2000.209
Bao, S.D. 2002. Soil agricultural chemical analysis, 3rd ed. Beijing: China Agricultural Press.
Blais, M., J.É. Tremblay, A.D. Jungblut, J. Gagnon, J. Martin, M. Thaler, and C. Lovejoy. 2012. Nitrogen fixation and identification of potential diazotrophs in the Canadian Arctic. Global Biogeochemical Cycles 26: GB3022. https://doi.org/10.1029/2011gb004096.
Bremmer, J.M., and C.S. Mulvaney. 1982. “Total nitrogen”, in methods of soil analysis—Part 2. In Chemical and microbiological properties, ed. C.A. Bluck, 595–624. Madison: American Society of Agronomy.
Carter, Martin R. 1993. Soil sampling and methods of analysis. Boca Raton: Lewis Publishers.
Coelho, Marcia R.R., Ivanildo E. Marriel, Sasha N. Jenkins, Clare V. Lanyon, Lucy Seldin, and Anthony G. O’Donnell. 2009. Molecular detection and quantification of nifH gene sequences in the rhizosphere of sorghum (Sorghum bicolor) sown with two levels of nitrogen fertilizer. Applied Soil Ecology 42: 48–53. https://doi.org/10.1016/J.APSOIL.2009.01.010.
Cole, James R., Qiong Wang, Jordan A. Fish, Benli Chai, Donna M. McGarrell, C. Yanni Sun, Titus Brown, Andrea Porras-Alfaro, Cheryl R. Kuske, and James M. Tiedje. 2014. Ribosomal database project: Data and tools for high throughput rRNA analysis. Nucleic Acids Research 42: D633–D642. https://doi.org/10.1093/nar/gkt1244.
Edgar, Robert C. 2013. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nature Methods 10: 996–998. https://doi.org/10.1038/nmeth.2604.
Edgar, Robert C., Brian J. Haas, Jose C. Clemente, Christopher Quince, and Rob Knight. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27: 2194–2200. https://doi.org/10.1093/bioinformatics/btr381.
Fadrosh, D.W., B. Ma, P. Gajer, N. Sengamalay, S. Ott, R.M. Brotman, and J. Ravel. 2014. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2: 6. https://doi.org/10.1186/2049-2618-2-6.
Gaby, John Christian, and Daniel H. Buckley. 2014. A comprehensive aligned nifH gene database: A multipurpose tool for studies of nitrogen-fixing bacteria. Database 2014: bau001. https://doi.org/10.1093/database/bau001.
Guan, S.Y., D. Zhang, and Z. Zhang. 1986. Soil enzyme and its research methods. Beijing: Agricultural.
Hammer, Ř., D.A.T. Harper, and P.D. Ryan. 2001. Past: Paleontological statistics software package for education and data analysis. Palaeontologia Electronica 4: 178.
Hardy, R.W., R.D. Holsten, E.K. Jackson, and R.C. Burns. 1968. The acetylene-ethylene assay for n(2) fixation: Laboratory and field evaluation. Plant Physiology 43: 1185–1207. https://doi.org/10.1104/pp.43.8.1185.
Heller, P., H.J. Tripp, K. Turk-Kubo, and J.P. Zehr. 2014. ARBitrator: A software pipeline for on-demand retrieval of auto-curated nifH sequences from GenBank. Bioinformatics 30: 2883–2890. https://doi.org/10.1093/bioinformatics/btu417.
Kaur, J., M. Verma, and R. Lal. 2011. Rhizobium rosettiformans sp. nov., isolated from a hexachlorocyclohexane dump site, and reclassification of Blastobacter aggregatus Hirsch and Muller 1986 as Rhizobium aggregatum comb. nov. International Journal of Systematic and Evolutionary Microbiology 61: 1218–1225. https://doi.org/10.1099/ijs.0.017491-0.
Kent, A.D., and E.W. Triplett. 2002. Microbial communities and their interactions in soil and rhizosphere ecosystems. Annual Review of Microbiology 56: 211–236. https://doi.org/10.1146/annurev.micro.56.012302.161120.
Li, C., Y. Dong, H. Li, J. Shen, and F. Zhang. 2016. Shift from complementarity to facilitation on P uptake by intercropped wheat neighboring with faba bean when available soil P is depleted. Scientific Reports 6: 18663. https://doi.org/10.1038/srep18663.
Li, Z.G., Y.M. Luo, and Y. Teng. 2008. Research methods for soil and environmental microbiology. Beijing: Science Press.
Li, X., P. Penttinen, Y. Gu, and X. Zhang. 2012. Diversity of nifH gene in rhizosphere and non-rhizosphere soil of tobacco in Panzhihua, China. Annals of Microbiology 62: 995–1001. https://doi.org/10.1007/s13213-011-0339-x.
Li, Y., H. Wen, L. Chen, and T. Yin. 2014. Succession of bacterial community structure and diversity in soil along a chronosequence of reclamation and re-vegetation on coal mine spoils in China. Edited by Daniele Daffonchio. PLoS ONE 9: e115024. https://doi.org/10.1371/journal.pone.0115024.
Li, Y.R., and L.T. Yang. 2015. Sugarcane agriculture and sugar industry in China. Sugar Tech 17: 1–8. https://doi.org/10.1007/s12355-014-0342-1.
Li, Y.R., X.Z. Zhou, and L.T. Yang. 2015. Biological nitrogen fixation in sugarcane and nitrogen transfer from sugarcane to cassava in an intercropping system. International Journal of Science and Nature 6: 214–218.
Magoc, T., and S.L. Salzberg. 2011. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27: 2957–2963. https://doi.org/10.1093/bioinformatics/btr507.
Orr, C.H., A. James, C. Leifert, J.M. Cooper, and S.P. Cummings. 2011. Diversity and activity of free-living nitrogen-fixing bacteria and total bacteria in organic and conventionally managed soils. Applied and Environmental Microbiology 77: 911–919. https://doi.org/10.1128/AEM.01250-10.
Perin, L., L. Martínez-Aguilar, G. Paredes-Valdez, J.I. Baldani, P. Estrada-de Los Santos, V.M. Reis, and J. Caballero-Mellado. 2006. Burkholderia silvatlantica sp. nov., a diazotrophic bacterium associated with sugarcane and maize. International Journal of Systematic and Evolutionary Microbiology 56: 1931–1937. https://doi.org/10.1099/ijs.0.64362-0.
Robertson, G.P., and P.M. Vitousek. 2009. Nitrogen in agriculture: balancing the cost of an essential resource. Annual Review of Environment and Resources 34: 97–125. https://doi.org/10.1146/annurev.environ.032108.105046.
Ruan, A., R. He, S. Xu, and T. Lin. 2009. Effect of dissolved oxygen on nitrogen purification of microbial ecosystem in sediments. Journal of Environmental Science and Health, Part A 44: 397–405. https://doi.org/10.1080/10934520802659778.
Sachs, J.L., S.W. Kembel, A.H. Lau, and E.L. Simms. 2009. In situ phylogenetic structure and diversity of wild Bradyrhizobium communities. Applied and Environmental Microbiology 75: 4727–4735. https://doi.org/10.1128/AEM.00667-09.
Solanki, M.K., F.Y. Wang, Z. Wang, C.N. Li, T.J. Lan, R.K. Singh, P. Singh, L.T. Yang, and Y.R. Li. 2018. Rhizospheric and endospheric diazotrophs mediated soil fertility intensification in sugarcane–legume intercropping systems. Journal of Soil and Sediments. https://doi.org/10.1007/s11368-018-2156-3.
Solanki, M.K., Z. Wang, F.Y. Wang, C.N. Li, T.J. Lan, R.K. Singh, P. Singh, L.T. Yang, and Y.R. Li. 2017. Intercropping in sugarcane cultivation influenced the soil properties and enhanced the diversity of vital diazotrophic bacteria. Sugar Tech 19: 136–147. https://doi.org/10.1007/s12355-016-0445-y.
Song, B., and B.B. Ward. 2003. Nitrite reductase genes in halobenzoate degrading denitrifying bacteria. FEMS Microbiology Ecology 43: 349–357. https://doi.org/10.1111/j.1574-6941.2003.tb01075.x.
Tai, X.S., W.L. Mao, G.X. Liu, T. Chen, W. Zhang, X.K. Wu, H.Z. Long, B.G. Zhang, and T.P. Gao. 2014. Distribution of ammonia oxidizers in relation to vegetation characteristics in the Qilian Mountains, northwestern China. Biogeosciences Discussions 11: 5123–5146. https://doi.org/10.5194/bgd-11-5123-2014.
Tai, X.S., W.L. Mao, G.X. Liu, T. Chen, W. Zhang, X.K. Wu, H.Z. Long, B.G. Zhang, and Y. Zhang. 2013. High diversity of nitrogen-fixing bacteria in the upper reaches of the Heihe River, northwestern China. Biogeosciences 10: 5589–5600. https://doi.org/10.5194/bg-10-5589-2013.
Walkley, A., and I.A. Black. 1934. An examination of the Degtjareff method for determining soil organic matter, and a proposed modification of the chromic acid titration method. Soil Science 37: 29–38.
Wang, Q., G.M. Garrity, J.M. Tiedje, and J.R. Cole. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology 73: 5261–5267. https://doi.org/10.1128/AEM.00062-07.
Wang, Z.G., X. Jin, X.G. Bao, X.F. Li, J.H. Zhao, J.H. Sun, P. Christie, and L. Li. 2014. Intercropping enhances productivity and maintains the most soil fertility properties relative to sole cropping. Edited by Ben Bond-Lamberty. PLoS ONE 9: e113984. https://doi.org/10.1371/journal.pone.0113984.
Xie, C.H., and A. Yokota. 2005. Reclassification of Alcaligenes latus strains IAM 12599T and IAM 12664 and Pseudomonas saccharophila as Azohydromonas lata gen. nov., comb. nov., Azohydromonas australica sp. nov. and Pelomonas saccharophila gen. nov., comb. nov., respectively. International Journal of Systematic and Evolutionary Microbiology 55: 2419–2425. https://doi.org/10.1099/ijs.0.63733-0.
Xie, C.H., and A. Yokota. 2006. Sphingomonas azotifigens sp. nov., a nitrogen-fixing bacterium isolated from the roots of Oryza sativa. International Journal of Systematic and Evolutionary Microbiology 56: 889–893. https://doi.org/10.1099/ijs.0.64056-0.
Yang, W., Z. Li, J. Wang, P. Wu, and Y. Zhang. 2013. Crop yield, nitrogen acquisition and sugarcane quality as affected by interspecific competition and nitrogen application. Field Crops Research 146: 44–50. https://doi.org/10.1016/J.FCR.2013.03.008.
Yousuf, B., R. Kumar, A. Mishra, and B. Jha. 2014. Differential distribution and abundance of diazotrophic bacterial communities across different soil niches using a gene-targeted clone library approach. FEMS Microbiology Letters 360: 117–125. https://doi.org/10.1111/1574-6968.12593.
Zehr, J.P., M.T. Mellon, and S. Zani. 1998. New nitrogen-fixing microorganisms detected in oligotrophic oceans by amplification of nitrogenase (nifH) genes. Applied and Environmental Microbiology 64: 3444–3450.
Zehr, J.P., B.D. Jenkins, S.M. Short, and G.F. Steward. 2003. Nitrogenase gene diversity and microbial community structure: a cross-system comparison. Environmental Microbiology 5(7): 539–554.
Zeng, Q., X. Wu, J. Wang, and X. Ding. 2017. Phosphate solubilization and gene expression of phosphate-solubilizing bacterium Burkholderia multivorans WS-FJ9 under different levels of soluble phosphate. Journal of Microbiology and Biotechnology 27: 844–855. https://doi.org/10.4014/jmb.1611.11057.
Zhang, L.H., and S.F. Chen. 2012. Pseudacidovorax intermedius NH-1, a novel marine nitrogen-fixing bacterium isolated from the South China Sea. World Journal of Microbiology & Biotechnology 28: 2839–2847. https://doi.org/10.1007/s11274-012-1093-3.
Zou, Y., J. Zhang, D. Yang, X. Chen, J. Zhao, W. Xiu, X. Lai, and G. Li. 2011. Effects of different land use patterns on nifH genetic diversity of soil nitrogen-fixing microbial communities in Leymus Chinensis steppe. Acta Ecologica Sinica 31: 150–156. https://doi.org/10.1016/J.CHNAES.2011.03.004.
Funding
Present research work supported by Grants from GXAAS (No. GNKB2014021); the Grants from the National High Technology Research and Development Program (“863” Program) of China (2013AA102604), National Natural Science Foundation of China (31171504, 31101122, 31471449), Guangxi Special Funds for Bagui Scholars’s and Distinguished Experts, Guangxi Natural Science Foundation and Guangxi Academy of Agriculture Sciences Fund (2011GXNSFF018002, 2012GXNSFDA053011, 2013NXNSFAA019073 and GuiNongKe2014YD01).
Author information
Authors and Affiliations
Contributions
YRL, LTY designed the study. MKS, FYW and CNL conducted the experiments. MKS and ZW did the isolation and characterization of microbes. TJL supported data collection and analysis. MKS, CNL, RKS, PS, LTY and YRL wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
There is no conflict of interest declared by all authors.
Ethical Statement
N/A
Informed Consent
N/A
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Solanki, M.K., Wang, FY., Li, CN. et al. Impact of Sugarcane–Legume Intercropping on Diazotrophic Microbiome. Sugar Tech 22, 52–64 (2020). https://doi.org/10.1007/s12355-019-00755-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12355-019-00755-4