
Abstract
Spinocerebellar ataxia (SCA17) is a rare genetic disorder characterized by a variety of neuropsychiatric symptoms. Recently, using magnetic resonance imaging (MRI) voxel-based morphometry (VBM), several specific functional–structural correlations comprising differential degeneration related to motor and psychiatric symptoms were reported in patients with SCA17. To investigate gray matter volume (GMV) changes over time and its association to clinical neuropsychiatric symptomatology, nine SCA17 mutation carriers and nine matched healthy individuals underwent a detailed neuropsychiatric clinical examination and a high-resolution T1-weighted volume MRI scan, both at baseline and follow-up after 18 months. Follow-up images revealed a progressive GMV reduction in specific degeneration patterns. In contrast to healthy controls, SCA17 patients showed a greater atrophy not only in cerebellar regions but also in cortical structures such as the limbic system (parahippocampus, cingulate) and parietal precuneus. Clinically, progression of motor symptoms was more pronounced than that of psychiatric symptoms. Correlation with the clinical motor scores revealed a progressive reduction of GMV in cerebellar and cerebral motor networks, whereas correlation with psychiatric scores displayed a more widespread GMV impairment in frontal, limbic, parietal, and also cerebellar structures. Interestingly, changes in global functioning were correlated with bilateral atrophy within the para-/hippocampus. While there was a good temporal association between worsening of motor symptoms and progression in cerebral and cortical neurodegeneration, the progression in psychiatric related neurodegeneration seemed to be more widespread and complex, showing progressive atrophy that preceded the further development of clinical psychiatric symptoms.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Spinocerebellar ataxia 17 (SCA17) is a rare, autosomal dominantly inherited neurodegenerative disorder characterized by cerebellar ataxia, epilepsy, extrapyramidal and pyramidal symptoms, and a wide spectrum of psychiatric disorders [1–8]. Examples for cognitive and behavioral abnormalities in SCA17 patients include cognitive impairment, dementia, adjustment disorder, personality changes, depression, phobia, and schizophrenia [2, 3, 7]. The disease-related mutation causes an expanded polyglutamine in the TATA-binding protein [8, 9]. The mean age of onset is about 30 years for individuals with full-penetrance alleles, but can range from 3 to 55 years [10]. The pathoanatomical and pathophysiological basis of the development of neuropsychiatric symptoms have not yet been well understood but the presence of cognitive and emotional symptoms of this neurodegenerative disorder has received increasing attention. Recently, we reported specific functional–structural correlations including differential patterns of degeneration related to motor and psychiatric signs in SCA17 [2]. The purpose of the present study was to capture the dynamics of gray matter volume (GMV) atrophy changes in the context of the clinical neuropsychiatric course of deterioration in these patients. We were interested in the question whether the progress of neuropsychiatric symptoms can be explained by progression of gray matter loss and if this was true, which brain structures would have been mostly affected by this progression. Structural imaging techniques such as voxel-based morphometry (VBM) are well suited to assess progression of GMV loss. Longitudinal magnetic resonance imaging (MRI) measurements suit best to assess structural changes over time against progression of neuropsychiatric signs of neurodegenerative disorders. To the best of our knowledge, such longitudinal structural studies have been lacking for SCA17 so far. Based on our recent systematic cross-sectional study [2], we had two major aims: Firstly, to compare GMV over an 18-month period between patients with SCA17 and healthy controls for the identification of changes specifically associated with neurodegenerative progression in SCA17. Secondly, we aimed at elucidating cerebral structures significantly involved in the progression of neuropsychiatric symptoms over time by correlating structural data with specific clinical neurological motor and psychiatric scales.
Methods
Subjects
SCA17 patients were recruited from the outpatient movement disorders clinics at the Departments of Neurology, Universities of Rostock and Luebeck, Germany, where the patients have been diagnosed and followed up on a regular basis. Nine carriers of a pathological repeat expansion in the SCA17 gene [7], seven male, two female (mean age: 40.1 (baseline)/41.6 (follow-up) years, standard deviation (SD): (± 11.1/11.0)), and nine age- and sex-matched healthy volunteers, seven male, two female, (mean age: 38.3 (baseline)/39.7 (follow-up) years, SD: (± 8.9/9.7)) were studied in 2005, and about 1.5 years later in 2007 (SCA17: 1.4 ± 0.1 years (mean ± SD), controls: 1.3 ± 0.7 years). Age differences between both groups were not significant (p = 0.73). All patients were interviewed and clinically examined to evaluate the neurological (C.K., J.H.) and psychiatric status (R.L., S.S.) using a standardized examination procedure at both times (International Cooperative Ataxia Rating Scale (ICARS), spasticity rating scale (in accordance with the Ashworth score), motor part of the Unified Parkinson's Disease Rating Scale (UPDRS-III), Mini-Mental State Examination (MMSE), personality change due to an organic brain disorder (F07), Global Assessment of Functioning (GAF). Details of these procedures have been described previously [2].
At baseline clinical assessment, seven of nine individuals with SCA17 mutations were markedly and two mildly affected. Most of our SCA17 patients clearly displayed cerebellar, extrapyramidal, and pyramidal signs, and these motor signs showed progress over time (Table 1).
In more detail, signs of ataxia were observed in eight at baseline and in nine SCA17 patients at follow-up. The phenotypic spectrum was wide ranging from mild to severe cerebellar signs with mean ICARS score of 24.9 (SD = 2.7) at baseline and 38.6 (SD = 4.0) at follow-up. Most of our patients showed mild signs of spasticity (Ashworth score at baseline: 1.1 ± 0.1 and follow-up: 1.2 ± 0.1). There was hardly any change in spasticity scores over the follow-up period. Within 18 months, there was a significant increase regarding the mean UPDRS-III scores (at baseline: 22.7 ± 2.5, at follow-up: 27.8 ± 2.5; Table 1). The mean MMSE percent rank was 76.9% ± 3.1 at baseline and 72.1% ± 3.8 at follow-up, ranging from 100% to 20%. There were no significant changes from baseline to follow-up for either personality change scores according to ICD-10 item F07 (baseline: 1.77 ± 0.2, follow-up: 2.0 ± 0.2) or GAF scores (baseline: 71.3 ± 3.4, follow-up: 67.5 ± 3.8).
Normal subjects were not related to the SCA17 subjects. The following exclusion criteria were used: a history of neurologic or psychiatric illnesses, prior exposure to neuroleptic agents or drug abuse, a medical history of hypertension, cardiovascular disease, or diabetes mellitus, and an abnormal neurologic examination. The study was approved by the local ethics committee, and written informed consent was obtained from all participating subjects in accordance with the Declaration of Helsinki [11] (http://www.wma.net/e/policy/b3.htm).
MRI Scanning
Morphometric Analysis
Scanning was performed with a 1.5 T whole-body scanner (Symphony, Siemens, Erlangen, Germany). All subjects underwent structural MRI imaging using a T1-weighted FLASH 3D MR sequence (echo time = 5 ms; repetition time = 5 ms; flip angle = 30°; isotropic voxel size 1 × 1 × 1 mm3).
Morphometric analysis was performed on a voxel-wise statistical approach using SPM2 software (Wellcome Department of Imaging Neuroscience, Institute of Neurology, UCL, London, www.fil.ion.ucl.ac.uk/spm) implemented in Matlab version 6.5 (Mathworks, Sherborn, MA, USA). Each image was normalized using the International Consortium for Brain Mapping template (Montreal Neurological Institute, Montreal, Canada), which approximates Talairach space. We applied a 12-parameter affine transformation to correct image size and position. Regional volumes were preserved, while corrections for global differences in whole brain volume were made. The normalized images of all subjects and patients were averaged and smoothed with a Gaussian kernel of 8 mm full width at half maximum (FWHM). Using the optimized procedure [12], these normalized images were employed to create a new template with reduced scanner- and population-specific bias. Each image was then locally deformed to the new template using a nonlinear spatial transformation [13], accounting for the remaining shape differences between the images and the template, and improving the overlap of corresponding anatomical structures. Finally, using a modified mixture model cluster analysis, normalized images were corrected for nonuniformities in signal intensity and partitioned into gray and white matter, Cerebrospinal fluid (CSF), and background. To remove unconnected nonbrain voxels, we applied a series of morphological erosions and dilations to the segmented images [12]. Gray matter images were smoothed with a Gaussian kernel of 12 mm FWHM. Using a general linear model, a voxel-by-voxel one-way analysis of variance was computed to detect differences in GMV between groups. For the avoidance of possible edge effects around the border between gray and white matter or CSF, an absolute gray matter threshold of 0.25 was used.
Statistical Design
Firstly, canonical contrasts were calculated between nine SCA17 patients and their corresponding group of control subjects for baseline and follow-up. To avoid potential artifacts due to global changes in brain size over time, the groups of controls that were also scanned at both time points were implemented in this study. To investigate changes over the course of time in two groups of subjects, a statistical design was performed in SPM2, containing the images of the two groups (SCA17 patients and controls) and two conditions per subject (baseline and follow-up). Secondly, a correlation between morphometric data and the clinical scores (ataxia, spasticity, UPDRS-III, MMSE, GAF, F07) was performed. For the comparison between baseline and follow-up, the respective clinical score was used as a covariate in the statistical design for the longitudinal data. Of note, the neuropsychiatric data for patient 9 are lacking for T2; therefore, the scan was not included in this design. Based on our previous morphometric results in SCA17 patients, we hypothesized a progressive GMV reduction in the previously reported regions, such as the cerebellum, basal ganglia, ventral striatum, limbic system, and parietal regions such as the precuneus [2]. For the statistical analysis, an explorative threshold of p < 0.01 (uncorrected) was used. Voxels were thresholded at a Z score >2.5, and clusters with a cluster extent (ke) >60 are reported. Region of interest analyses using an anatomical mask were also employed when having a strict a priori hypothesis to an anatomically restricted area of expected changes in GMV (e.g., regions that were identified in the cross-sectional analysis at baseline). The WFU PickAtlas [14] and the Schmahmann MRI Atlas of the Human Cerebellum [15] were used as an anatomical reference to assess the exact localization of significantly atrophic gray matter areas.
Results
Categorical Comparison
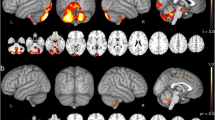
At baseline (T1) and follow-up (T2), a general GMV decrease was found in the cerebellum, the limbic lobe as well as in frontotemporoparietal structures in SCA17 patients compared to healthy participants. The most prominent GMV loss in SCA17 patients was indeed found in the cerebellum at baseline and also at follow-up. Moreover, large regions with reduced GMV were observed in inferior frontal and limbic structures, basal ganglia, and temporoparietal structures. Figure 1 and Table 2 show the longitudinal findings contrasting the baseline and follow-up assessments (T1 > T2) between SCA17 patients and controls. One of the most prominent findings besides cerebellar atrophy was a GMV decrease in the limbic uncus representing a part of the parahippocampal gyrus that was accentuated on the left side. Similar to previous findings [2], the results showed a GMV decrease in the bilateral parietal precuneus and the cingulate (Table 2).

Longitudinal findings contrasting the baseline and follow-up (T1 > T2) assessments between SCA17 patients and healthy controls. Gray matter volume atrophy was found bilaterally in cerebellum, inferior frontal, and limbic structures such as parahippocampus. The overlay maps are superimposed and rendered on the SPM standard T1-weighted template (p < 0.05). The color bar represents the T values
Clinical Correlations
Ataxia
Figure 2a illustrates the longitudinal regression analysis (T1 > T2) with the ataxia ICARS scores, comprising the contrast between baseline and follow-up in SCA17 patients. At this, the right cerebellar posterior lobe (lobule VIII) was most affected, whereas the left side (lobule VIII) and the left cerebellar anterior lobe (lobule V) were impaired to a lesser extent (Table 3).

a The regression analysis with the ICARS scores over time (T1 > T2) revealed a significant gray matter volume loss in left anterior cerebellar lobe and bilateral posterior cerebellar lobe. b Within the UPDRS motor scores the analysis displayed a gray matter volume decrease mainly in the left putamen. c Longitudinal changes of global functioning (GAF score) over follow-up were correlated with reductions of gray matter in hippo- and parahippocampus bilaterally, but also with widespread atrophy in frontoparietal structures including precuneus, cingulate, and cerebellum. The overlay maps are superimposed and rendered on the SPM standard T1-weighted template (p < 0.05). The color bar represents the T values
Spasticity
The longitudinal approach (T1 > T2) revealed besides a decrease in GMV in the cerebellum no significant changes in GMV (Table 3).
UPDRS-III
Longitudinally, the contrast between baseline and follow-up revealed a GMV decrease mainly in the left putamen (Table 3, Fig. 2b).
MMSE
Correlating the morphometric data with the MMSE scores, the contrast (T1 > T2) revealed a decrease in GMV in ventral striatum. Besides this finding, GMV was decreased in left parietal precuneus and right cerebellum posterior lobe (Table 3).
F07
Although there was hardly any change in clinical signs, the contrast between baseline and follow-up revealed a bilateral neurodegenerative progression of GMV in left middle orbitofrontal gyrus, right middle frontal gyrus, and left anterior cerebellar lobe (Table 3).
GAF
The contrast (T1 > T2) displayed a GMV in the hippocampus and parahippocampus region but also a widespread decrease in GMV in the left superior frontal gyrus (BA6), the bilateral cingulate cortex, in parietal structures such as the right precuneus and the right superior parietal lobule, and the bilateral cerebellum (Table 3, Fig. 2c).
Discussion
This is the first systematic longitudinal study on individuals carrying an SCA17 mutation. We used VBM to evaluate brain tissue changes over an 18-month period in these individuals and compared their results with those of a healthy group that was followed up over the same time interval. Morphometric changes were related to the clinical progression of neuropsychiatric symptoms.
The major findings are: Firstly, in SCA17 patients, we found a progressive GMV atrophy mainly in the cerebellum, cerebral motor networks, parietal structures, and the limbic system. Secondly, there was a good temporal association between the deterioration in motor symptoms and the progression in cerebellar and cortical neurodegeneration, whereas progression in GMV loss in frontal, parietal, limbic, and cerebellar structures seemed to precede clinical progression of more psychiatric symptoms such as personality change and global functioning.
SCA17 is a rapidly progressive neurodegenerative disease when compared to other neurodegenerative disorders such as Parkinson's disease. Thus, we assumed that a follow-up interval of 18 months would be long enough to capture the clinical and structural progression in SCA17. Once the disease has manifested, unremitting progression of gray matter loss results in deterioration of neuropsychiatric impairment with death occurring at a mean age 39 ± 20 years after a mean disease duration of 19 ± 9 years [16]. VBM is perfectly suited to visualize brain shape differences between groups, changes over time related to the effects of neuropsychiatric symptoms on the brain, and to highlight patterns of altered brain structure related to these conditions. We confirmed our previous work in the localization of specific patterns of cerebral degeneration and extended the longitudinal part in order to capture the dynamics of GMV atrophy associated with the main neuropsychiatric features.
Categorical Comparison over Time
In SCA17 patients, VBM revealed a significant GMV loss over time in contrast to the controls. As we expected, cerebellar structures, i.e., its left anterior lobe and right posterior lobe, were most atrophic. Moreover, our data clearly demonstrate that neurogenerative processes also affect cerebral structures such as left limbic cingulate, parahippocampus bilaterally, and parietal precuneus. These findings comply well with the cross-sectional patterns in the previously described larger population of SCA17 patients [2] and underline the ongoing progression of neurodegeneration in the whole brain.
Regression Analysis between GMV Degeneration and Motor Scales over Time
The main finding was indeed the progression in atrophy in the cerebellum. It is well known that the cerebellum is the main target of neurodegeneration in SCA. On the structural level, a cerebellar atrophy was demonstrated in conventional structural neuroimaging in SCA17 [7, 17] and also in other SCA genotypes, such as SCA1, 2, and 3 [18]. On the metabolic level, in SCA1, 2, 3, and 6 cerebellar glucose metabolic deficits were reported using fluorodeoxyglucose positron emission tomography (FDG PET) [19, 20].
The basal ganglia, playing a fundamental role in the extrapyramidal motor system, seem to be involved in the pathological process in SCA17. Striatal dysfunction in SCA17 has been demonstrated in several ways: T2-weighted MRI displayed putaminal rim hyperintensity [21]. FDG PET revealed a reduced striatal glucose metabolism [22–24], and single photon emission tomography (SPECT) scans demonstrated a progressive marked reduction of striatal dopamine transporter availability over a 2-year follow-up period [23, 24]. Using FP-CIT SPECT, it was demonstrated that nigrostriatal dysfunction does not occur in the earliest disease stages and correlates with the clinical severity of ataxia in manifesting patients with a fully developed phenotype [25]. The progressive decrease in the basal ganglia over time and in relation to extrapyramidal signs may therefore provide the pathological substrate to these metabolic observations.
Regression Analysis between GMV Degeneration and Psychiatric Scales over Time
The degree of the personality change and global functioning due to SCA17 mutation related brain dysfunction were both correlated with brain volume reduction in a wide range of cortical areas including frontal, parietal (precuneus), limbic (anterior and posterior cingulate, parahippocampus), and cerebellar structures. While the correlation with the degree of personality change after follow-up only revealed some marginal changes in frontal and cerebellar structures, this was different for global psychosocial functioning. The extent of gray matter loss in frontal, limbic, and cerebellar degeneration that was correlated with GAF scores was found to increase during follow-up of 18 months. The frontal and the limbic systems represent the main neuroanatomical structures for the emotional system [26], whereas the precuneus appears to play an important role in fundamental cognitive functioning in humans. Anatomical studies of cytoarchitecture and connectivity have partially exposed the neural system to which the precuneus belongs, a widespread network of higher association cortical and subcortical structures, indicating the complexity of its behavioral specializations [27]. Recent functional imaging findings in healthy subjects have shown a central role for the precuneus in a wide spectrum of highly integrated tasks [27]. Thus, based on the involvement of precuneus and cingulate in attentional, motivational, and emotional processing [28] on the one hand, and the control of emotional behavior by frontal areas, the dysfunction within this circuit may explain a reduced ability to persevere with goal-directed activities and increased emotional irritability as being observed in most of our patients. Also disinhibited expression of needs or impulses and apathy may reflect frontal dysfunctions [2].
The finding of additional involvement of para- and hippocampal regions underlines its fundamental role in the neurodegenerative progression in SCA17 patients. Atrophy of the hippocampus has been described in a variety of psychiatric disorders [29–31]. There is evidence that hippocampal atrophy even precedes the development of psychiatric symptoms although the exact role and basis for that are still unknown [32, 33]. Hippocampal degeneration may reflect the high accumulation of dementia in SCA17 patients as reported previously [2]. Otherwise, the parahippocampal region, an important part of the limbic system, contains various and complex interconnections with regions that are involved in emotional processing.
The regression analyses with personality change and global functioning also showed correlations with the cerebellum, known to be involved in nonmotor functions. Based on neuroanatomical studies demonstrating a variety of complex connections between the cerebellum and frontal and limbic areas [34], basal ganglia and other cortical structures [35], functional studies [36], and reports of patients with cerebellar diseases (e.g., [37, 38]), point to an important modulating role of the cerebellum for cognition and emotion processing. The disruption of these cerebrocerebellar circuits may result in the observed psychiatric symptoms.
Based on the important role of the ventral striatum, in particular the nucleus accumbens, for the transition from “emotion and motivation to action,” the high correlation between the MMSE scores and the bilateral ventral striatal atrophy may explain reduced capability of emotional and behavioral control as observed in SCA17 patients suffering from dementia [2].
The discrepancy between progression over time in frontal, limbic, parietal, and cerebellar structures but very mild clinical psychiatric symptom progression is in contrast to the consistency of motor symptom progression and increased GMV atrophy in motor system-related areas during follow-up. The broad phenotypic spectrum in SCA17 seems to progress in variable stages with motor symptoms being usually more prominent whereas neuropsychiatric abnormalities may harder to define both for patients and physicians. The progression of structural alterations related to psychiatric disorders seems to be more complex and preceded the manifestation of clinical psychiatric symptom progression. Also, we have to consider that our sample size was small and included patients of various disease stages such as five patients on a high function level and four on a low function level. A deterioration of neuropsychiatric function during follow-up was only observed in the latter patient group.
Summary
The findings in this first longitudinal VBM study in SCA17 patients confirm previous cross-sectional analyses and revealed progressive GMV loss in specific degeneration patterns related to structure and function [2]. The progression at the structural level seems to mirror the progression of clinical motor features. The progression of structural findings related to psychiatric symptoms seems to be more distributed and interdependent. The ability to longitudinally evaluate GMV changes offers new opportunities for a better understanding of pathophysiological and pathoanatomical processes in SCA17 and may thus be helpful in monitoring novel treatment approaches for individuals carrying a SCA17 mutation.
References
Hagenah J, Reetz K, Zuhlke C, Rolfs A, Binkofski F, Klein C (2007) Predominant dystonia with marked cerebellar atrophy: a rare phenotype in familial dystonia. Neurology 68(24):2157 author reply -8
Lasek K, Lencer R, Gaser C, Hagenah J, Walter U, Wolters A et al (2006) Morphological basis for the spectrum of clinical deficits in spinocerebellar ataxia 17 (SCA17). Brain 129(Pt 9):2341–2352
Maltecca F, Filla A, Castaldo I, Coppola G, Fragassi NA, Carella M et al (2003) Intergenerational instability and marked anticipation in SCA-17. Neurology 61(10):1441–1443
Hagenah JM, Zuhlke C, Hellenbroich Y, Heide W, Klein C (2004) Focal dystonia as a presenting sign of spinocerebellar ataxia 17. Mov Disord 19(2):217–220
Hernandez D, Hanson M, Singleton A, Gwinn-Hardy K, Freeman J, Ravina B et al (2003) Mutation at the SCA17 locus is not a common cause of Parkinsonism. Parkinsonism Relat Disord 9(6):317–320
Zuhlke C, Dalski A, Schwinger E, Finckh U (2005) Spinocerebellar ataxia type 17: report of a family with reduced penetrance of an unstable Gln49 TBP allele, haplotype analysis supporting a founder effect for unstable alleles and comparative analysis of SCA17 genotypes. BMC Med Genet 6(1):27
Rolfs A, Koeppen AH, Bauer I, Bauer P, Buhlmann S, Topka H et al (2003) Clinical features and neuropathology of autosomal dominant spinocerebellar ataxia (SCA17). Ann Neurol 54(3):367–375
Nakamura K, Jeong SY, Uchihara T, Anno M, Nagashima K, Nagashima T et al (2001) SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet 10(14):1441–1448
Koide R, Kobayashi S, Shimohata T, Ikeuchi T, Maruyama M, Saito M et al (1999) A neurological disease caused by an expanded CAG trinucleotide repeat in the TATA-binding protein gene: a new polyglutamine disease? Hum Mol Genet 8(11):2047–2053
Zuhlke C, Burk K (2007) Spinocerebellar ataxia type 17 is caused by mutations in the TATA-box binding protein. Cerebellum:1–8
Helsinki WMADo (2000) Ethical principles for medical research involving human subjects. JAMA 284(23):3043–3045
Good CD, Johnsrude IS, Ashburner J, Henson RN, Friston KJ, Frackowiak RS (2001) A voxel-based morphometric study of ageing in 465 normal adult human brains. Neuroimage 14(1 Pt 1):21–36
Ashburner J, Friston KJ (1999) Nonlinear spatial normalization using basis functions. Hum Brain Mapp 7(4):254–266
Maldjian JA, Laurienti PJ, Kraft RA, Burdette JH (2003) An automated method for neuroanatomic and cytoarchitectonic atlas-based interrogation of fMRI data sets. Neuroimage 19(3):1233–1239
Schmahmann JD, Doyon J, Toga AW, Terides M, Evans AC (2000) MRI Atlas of the human cerebellum. Academic, San Diego, California
Wells R, Ashizawa T (2006) Spinocerebellar Ataxia 17 and Huntington's disease-like 4. Genetic instabilities, neurological diseases. Second Edition. Academic Press
Toyoshima Y, Yamada M, Onodera O, Shimohata M, Inenaga C, Fujita N et al (2004) SCA17 homozygote showing Huntington's disease-like phenotype. Ann Neurol 55(2):281–286
Klockgether T, Skalej M, Wedekind D, Luft AR, Welte D, Schulz JB et al (1998) Autosomal dominant cerebellar ataxia type I. MRI-based volumetry of posterior fossa structures and basal ganglia in spinocerebellar ataxia types 1, 2 and 3. Brain 121(Pt 9):1687–1693
Wang PS, Liu RS, Yang BH, Soong BW (2007) Regional patterns of cerebral glucose metabolism in spinocerebellar ataxia type 2, 3 and 6: a voxel-based FDG-positron emission tomography analysis. J Neurol 254(7):838–845
Wullner U, Reimold M, Abele M, Burk K, Minnerop M, Dohmen BM et al (2005) Dopamine transporter positron emission tomography in spinocerebellar ataxias type 1, 2, 3, and 6. Arch Neurol 62(8):1280–1285
Loy CT, Sweeney MG, Davis MB, Wills AJ, Sawle GV, Lees AJ et al (2005) Spinocerebellar ataxia type 17: extension of phenotype with putaminal rim hyperintensity on magnetic resonance imaging. Mov Disord
Lin IS, Wu RM, Lee-Chen GJ, Shan DE, Gwinn-Hardy K (2007) The SCA17 phenotype can include features of MSA-C, PSP and cognitive impairment. Parkinsonism Relat Disord 13(4):246–249
Gunther P, Storch A, Schwarz J, Sabri O, Steinbach P, Wagner A et al (2004) Basal ganglia involvement of a patient with SCA 17—a new form of autosomal dominant spinocerebellar ataxia. J Neurol 251(7):896–897
Minnerop M, Joe A, Lutz M, Bauer P, Urbach H, Helmstaedter C et al (2005) Putamen dopamine transporter and glucose metabolism are reduced in SCA17. Ann Neurol 58(3):490–491
Salvatore E, Varrone A, Sansone V, Nolano M, Bruni AC, De Rosa A et al (2006) Characterization of nigrostriatal dysfunction in spinocerebellar ataxia 17. Mov Disord 21(6):872–875
Heinzel A, Bermpohl F, Niese R, Pfennig A, Pascual-Leone A, Schlaug G et al (2005) How do we modulate our emotions? Parametric fMRI reveals cortical midline structures as regions specifically involved in the processing of emotional valences. Brain Res Cogn Brain Res 25(1):348–358
Cavanna AE, Trimble MR (2006) The precuneus: a review of its functional anatomy and behavioural correlates. Brain 129(Pt 3):564–583
Davis KD, Taylor KS, Hutchison WD, Dostrovsky JO, McAndrews MP, Richter EO et al (2005) Human anterior cingulate cortex neurons encode cognitive and emotional demands. J Neurosci 25(37):8402–8406
Kantarci K, Jack CR Jr (2003) Neuroimaging in Alzheimer disease: an evidence-based review. Neuroimaging Clin N Am 13(2):197–209
Frodl T, Schaub A, Banac S, Charypar M, Jager M, Kummler P et al (2006) Reduced hippocampal volume correlates with executive dysfunctioning in major depression. J Psychiatry Neurosci 31(5):316–323
Reetz K, Lencer R, Steinlechner S, Gaser C, Hagenah J, Buchel C et al (2008) Limbic and frontal cortical degeneration is associated with psychiatric symptoms in PINK1 mutation carriers. Biol Psychiatry
Dhikav V, Anand KS (2007) Is hippocampal atrophy a future drug target? Med Hypotheses 68(6):1300–1306
McEwen BS (1997) Possible mechanisms for atrophy of the human hippocampus. Mol Psychiatry 2(3):255–262
Middleton FA, Strick PL (2000) Basal ganglia and cerebellar loops: motor and cognitive circuits. Brain Res Brain Res Rev 31(2–3):236–250
Alexander GE, Crutcher MD, DeLong MR (1990) Basal ganglia-thalamocortical circuits: parallel substrates for motor, oculomotor, "prefrontal" and "limbic" functions. Prog Brain Res 85:119–146
Liotti M, Mayberg HS, Brannan SK, McGinnis S, Jerabek P, Fox PT (2000) Differential limbic–cortical correlates of sadness and anxiety in healthy subjects: implications for affective disorders. Biol Psychiatry 48(1):30–42
Schmahmann JD, Sherman JC (1997) Cerebellar cognitive affective syndrome. Int Rev Neurobiol 41:433–440
Leroi I, O'Hearn E, Marsh L, Lyketsos CG, Rosenblatt A, Ross CA et al (2002) Psychopathology in patients with degenerative cerebellar diseases: a comparison to Huntington's disease. Am J Psychiatry 159(8):1306–1314
Acknowledgments
We gratefully thank all the participants for their invaluable collaboration.
This work was supported by intramural grants from the Medical Faculty, University of Luebeck to KR (E06-2008) and CK. CK was further supported by the Hermann and Lilly Schilling Foundation. The Volkswagen Stiftung supported FB and CK.
Competing interests
The authors have no financial or other interests to disclose.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Reetz, K., Lencer, R., Hagenah, J.M. et al. Structural Changes Associated with Progression of Motor Deficits in Spinocerebellar Ataxia 17. Cerebellum 9, 210–217 (2010). https://doi.org/10.1007/s12311-009-0150-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12311-009-0150-4