
Abstract
Creutzfeldt–Jakob Disease (CJD) is characterized by bilateral basal ganglia hyperintensities on T2W and diffusion-weighted imaging (DWI) magnetic resonance imaging (MRI) scans, consistent with its extrapyramidal neurological manifestations. MRI is diagnostically uninformative about the cerebellar symptoms, equally prominent in CJD. This study was undertaken to explain this apparent paradox. Eleven CJD patients with definite cerebellar or brain stem symptoms were selected from a large prospective study, as well as 11 healthy controls matched for age and gender. All subjects participated in a standardized MRI protocol, including SPGR, fluid-attenuated inversion recovery (FLAIR), DWI and diffusion tensor imaging (DTI). All subjects underwent detailed examination by a neurologist blinded to the radiological findings, who predicted the expected site of cerebral abnormalities. MRI showed good sensitivity for the abnormalities predicted in the cortex (80–90%) and basal ganglia (100%). None of the standard MRI sequences, including DWI, DTI, and FLAIR, revealed any tissue abnormalities in cerebellum or brain stem. Apparent diffusion coefficient (ADC) values, however, were substantially and significantly elevated in several cerebellar structures, where also the volumetric (VBM) analysis revealed elevated cerebrospinal fluid volume, suggesting focal cerebellar atrophy in these CJD patients. In patients with CJD, DWI appears sensitive to the reduced diffusivity in cortex and basal ganglia but insensitive to cerebellar involvement. We propose that the radiological hallmark of cerebellar pathology in CJD is atrophy, revealed quantitatively by both VBM and elevated diffusivity, which is identifiable on ADC maps but poorly visualized in nonquantitative DWI images.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Creutzfeldt-Jakob Disease (CJD) [1] is the most common human prion disease, characterized by a rapidly progressive multifocal neurological dysfunction, myoclonic jerks, and a terminal state of global severe cognitive impairment. CJD can be etiologically classified into infectious, sporadic and familial forms. The familial forms are all caused by a mutation in the PrP gene, and the most prevalent form is found in Libyan Jews [2], caused by a Glu to Lys substitution in codon 200 (E200K). The disease usually affects the cerebral and cerebellar cortices as well as the basal ganglia, thalamus,] and brainstem. The main neuropathological features are spongiform degeneration, neuronal loss, astrocytosis, and cerebral accumulation of the abnormal prion protein, PrPSc. Spongiform changes are characteristically seen in cerebral cortex, with variable distribution and severity. Basal ganglia, thalamus, cerebellum, and brainstem are also variably involved [3, 4]. The cerebral distribution of the abnormal protein was reported to be primarily cortical, with significant concentrations also in basal ganglia and cerebellum [5, 6] A recent review [7] reported that the highest amounts of PrPSc were found in all four cortical lobes, entorhinal cortex, striatum, thalamus, and cerebellum, in the most common phenotype, but emphasized the phenotypic heterogeneity, which appears to depend on the interaction between the prion strain and the host genotype. PrPSc plaques are most common in the cerebellum [6].
Recently, it became evident that magnetic resonance imaging (MRI) imaging of the brain may have an important role in the diagnosis of the disease [8]. Typical MRI findings in CJD patients consist of hyperintense signals in the cortical ribbon, basal ganglia, and the thalamus on diffusion-weighted images (DWI) and fluid-attenuated inversion recovery (FLAIR). Despite the intense neuropathological phenotypic involvement of the cerebellum in CJD [9] and the high prevalence of clinical cerebellar symptoms [5, 10], it is typically negative in imaging studies [8, 11], even when the most sensitive modern modalities, (diffusion-weighted or FLAIR MRI) are used.
Thus, neuropathological changes occur in the cerebellum, as well as the cerebrum, and clinical signs and symptoms arise from both, but MRI is “blind” to the disease in the cerebellum. This discrepancy has not been systematically investigated with modern MRI methods. The heterogeneity of CJD may be relevant: it is possible that previous imaging studies happened to include patients without prominent cerebellar symptomatology. It is also possible that the most sensitive MRI sequences have not been tried on patients with cerebellar syndromes. To address this issue, we specifically investigated the MRI appearance of CJD patients with clear cerebellar signs and utilized quantitative MRI methods that have not been previously attempted.
Methods
Patients
The study group included 11 early-stage CJD patients (six men, five women, mean age 61 ± 7 years, range 52–75), selected on the basis of cerebellar or brainstem clinical presentation. The clinical diagnosis of CJD required a relatively rapid decline of cognitive function together with subacute changes in behavior and neurological deficits with at least two of the following features: myoclonus, visual or cerebellar symptoms, pyramidal, or extrapyramidal dysfunction, or akinetic mutism [12].
While we did not obtain neuropathological examinations, our diagnoses were confirmed by genetic information and clinical follow-up. Electroencephalogram showed periodic activity compatible with CJD in four patients, triphasic waves in one patient, and nonspecific slowing in six patients. Cerebrospinal fluid Tau protein levels were available in nine patients and were all elevated ranging between 789 and 1,277 pg/ml. Genetic testing revealed the presence of the E200K mutation in nine patients, while two patients had sporadic disease. All patients were followed to death, as part of a longitudinal prospective study. Disease severity was quantified by the CJD Rating Scale (CRS) [13], recently developed for this purpose.
MRI scanning was performed, on the average, 3.5 ± 2.7 months (range 1–7) from onset of the disease; at this time, the mean CRS score was 9 ± 3. The Mini-Mental Status (MMS) [14] was evaluated in seven patients, and yielded a mean score of 23 ± 6 (range 11–29). Survival (from onset to death) was 6.4 ± 4.3 months (range 2–13 months). Control subjects were healthy members of the patients' families (six men, five women, mean age 60 ± 10, range 46–80), who underwent the same diagnostic and imaging procedures as the patients. Their CRS scores were 0.45 ± 0.63, and their MMS 28 ± 2.
Imaging
MRI scans were performed on a 1.5T GE Signa system using a standard quadrature head coil. Sequences included a rapid scout, followed by DWI using a single-shot echo-planar spin echo sequence acquired in the axial plane with parameters of 5,000/85 (TR/TE), a field of view of 24 cm, an imaging matrix of 128 × 128, with b values of 0 s/mm2 (T2-weighted image) and 1,000 s/mm2 along three orthogonal directions, and 3-mm contiguous slices, resulting in 48 slices. A fluid-attenuated inversion recovery sequence had parameters of 9,000/110/2,200 (TR/TE/TI), and a T2-weighted fast spin echo (FSE) sequence had parameters of 4,000/90 (TR/TE) and an echo train length of 9. Both the FLAIR and FSE sequences were acquired in the axial plane and had a field of view of 22 cm, an imaging matrix of 256 × 256, 3-mm-thick slices with a 3-mm gap and 24 slices that were positioned so that their centers coincided with every other DWI slice. The imaging parameters in the high resolution T1-weighted sequence(SPGR) were 104 contiguous axial slices, FOV = 240 mm, TR/TE/FA 28 ms/6 ms//40°, 1.50 mm slice thickness with acquisition matrix 256 × 256, yielding reconstructed voxel dimensions at 0.94 × 0.094 × 1.50 mm. Finally, diffusion-tensor imaging (DTI) using a single-shot echo-planar spin echo sequence was acquired in the axial plane with parameters of 8,300/92.6 (TR/TE), a field of view of 26 cm, an imaging matrix of 128 × 128, with b values of 0 s/mm2 (T2-weighted image) and 1,000 s/mm2 along 25 directions, and 5 mm contiguous slices resulting in 26 slices.
The FLAIR, DTI and other sequences were used for clinical evaluation and lesion localization. The DWI and SPGR sequences were utilized for quantitative analyses, detailed below.
Quantitative Image Analysis
Because diffusion MRI is considered the most sensitive for CJD but the clinically used diffusion images are nonquantitative, diffusion quantification was our first step. The acquired DWI images were analyzed with a Matlab (MathWorks, Natick, MA) software package developed in house to calculate the apparent diffusion coefficient (ADC). Parenchymal regions were first delineated by applying the Brain Extraction Tool (BET, FSL 4.0, http://www.fmrib.ox.ac.uk/fsl/) to the b = 0 image. ADC was calculated by the standard Stejskal and Tanner [15] equation in each voxel:
where SI(b = 0) and SI(b = 1,000) are the signal intensities of diffusion-weighted images with applied b values of b1 = 0 s/mm2 and b2 = 1,000 s/mm2. Data quality was assured by visually inspecting each slice for any signal drop out in DWI, and if detected, the signal was rescaled to match its adjacent slices.
Voxel-wise analyses were processed by SPM5 (Wellcome Department of Cognitive Neurology, London). In the spatial processing step, the T2W (b = 0 s/mm2) image was first coregistered with the anatomical image (SPGR) using the mutual information algorithm, and the derived coregistration parameters were applied onto the computed ADC map. Normalization parameters were then derived by segmenting the SPGR T1W scan using the default values: 25 mm nonlinear warp frequency cutoff and canonical template of ICBM452 tissue probability maps [16, 17]. The normalization parameters derived from the SPGR scan were applied onto the ADC maps, which were also resliced at 2 × 2 × 2 mm and smoothed by an isotropic 4 mm Gaussian smoothing kernel.
The resulting smoothed and normalized images were submitted to a random effects analysis of covariance (ANCOVA) between controls and patients, with age as a covariate. A custom explicit mask was also created to eliminate all voxels likely to be pure cerebrospinal fluid (CSF) by setting the upper limit of ADC at 2,500 μm2/s. In each voxel, if any of the subjects' ADC value exceeds the upper limit, the voxel was excluded from analysis. Significance was defined as p < 0.001 (uncorrected) and k = 5.
Second, because the ADC data suggested novel atrophy findings in the cerebellum, we quantified the cerebrospinal fluid volumes and compared them between the controls and the patients in two ways: voxel based morphometry and volume averaged CSF volumes. For the VBM analysis [18], SPGR images were normalized and segmented using the default values as described earlier. Because images are warped during this step, the Jacobian determinant was derived and applied onto each voxel to account for volumetric change during normalization. Segmented and modulated images were then smoothed by an 8-mm Gaussian smoothing kernel and were submitted to random effects ANCOVA between controls and patients with age and total intracranial volume as covariates. An explicit mask was created by setting the lower limit of segmented image intensity at 0.05. Significance was defined as p < 0.005 (uncorrected) and k = 5. Volume averaged CSF volumes were also computed in each cluster found to be significant in the ADC voxel-wise analysis. In each cluster, voxel coordinates and corresponding CSF volumes were extracted from modulated and unsmoothed CSF image intensity.
Clinical Evaluations
All MRI scans were read by one neuroradiologist (CH) experienced in the evaluation of CJD, who was aware of the CJD diagnosis of the patients, but blind to the detailed neurological findings. The mean DWI image derived from DTI was primarily used for radiological localization, but all other sequences were also utilized. Scans were primarily read for the location of abnormalities, especially hyperintense signals in diffusion and T2W images.
All subjects underwent a structured neurological examination, based on a new scale for the symptoms of CJD [13] by neurologists blinded to the radiological findings. Based on these examinations, another neurologist (OC), also blind to radiological findings, predicted the expected site of cerebral abnormalities. The presence of frontal release signs predicted a frontal lobe lesion. Basal ganglia signs included rigidity, rest tremor or dystonia. Motor cortex involvement was suspected when hemiparesis or pyramidal signs were elicited, and the presence of ataxia, dysmetria, nystagmus, or dysarthria/dysphonia predicted cerebellar or brainstem involvement respectively [19].
Results
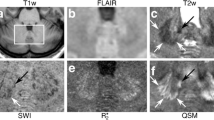
Frontal release signs were found in nine of 11 patients. Motor cortex abnormalities, including hemiparesis or pyramidal signs, were also found in nine of 11 patients. Basal ganglia signs, including rigidity, rest tremor or dystonia were evident in eight of 11 patients. Signs attributable to the brainstem (dysarthria/dysphonia) were found in seven of 11 patients. All 11 patients revealed clear cerebellar signs (ataxia, dysmetria, nystagmus), since this was the basis of their selection. When correlated with the clinical findings, MRI displayed definite signal abnormalities for the frontal cortex in eight of nine patients and for the motor cortex in seven of nine patients (Table 1). In the basal ganglia, the MRI was positive in ten patients, eight of whom had relevant clinical signs. None of the patients demonstrated brain stem or cerebellar MRI abnormalities. An illustrative case (FLAIR and DTI-derived DWI) is shown in Fig. 1, depicting strong basal ganglia and cortical abnormalities with no cerebellar findings.

Axial FLAIR (a–c) and DWI (d–f, derived from DTI) images of a typical CJD patient. This was a 60-year-old woman (case 2014) with significant pyramidal and cerebellar symptomatology, including limb and trunk ataxia, bradykinesia, and a positive snout reflex. MRI was obtained 6 months after initial onset, and she died 7 months later. Note the typically bright caudate, putamen, and parts of the thalamus, as well as cortical hyperintensity, mainly in temporal neocortex and insula. No significant cerebellar hyperintensity could be found.
Quantitative ADC analysis documented the expected diffusion reductions in the basal ganglia (Table 2). These were small, focal bilateral areas in the thalamus and lentiform nucleus, with about 15% reductions of diffusion; no other areas of the brain showed significant reductions. In contrast, there were many areas demonstrating significant elevations of diffusion. These areas included several cerebellar clusters, as well as many cortical areas (the largest clusters occurred in middle frontal gyrus and superior temporal gyrus). Any voxel where ADC values exceeded 2,500 μm2/s was excluded from analysis; in the included voxels, ADC elevations ranged from 23% to 49%.
VBM analysis demonstrated no CSF elevations in the basal ganglia. In contrast, CSF volume was significantly higher in two cerebellar locations that also showed higher ADC (Table 2). The full SPM analysis results for both ADC and CSF increases is shown in Fig. 2 to illustrate their spatial overlap.

The larger cerebellar loci of significantly elevated ADC (left) and CSF volume (right) in CJD patients are found bilaterally in the cerebellar nodule, shown here in three orthogonal projections. Peaks for both clusters are located at MNI coordinates x–8 y–54 z–36.
Further, we extracted CSF volumes (from the VBM analysis) for all four cerebellar clusters that were found to have significantly elevated ADC values. The mean CSF volumes from each subject were then subjected to a repeated-measures multivariate analysis of covariance, using diagnosis (CJD vs control) as grouping factor, the four clusters as repeated dependent measures, and age (with interactions) as a covariate. The effect of diagnosis was significant (F 1,18 = 11.53, p < 0.005), as was the effect of age (F 1,18 = 6.44, p < 0.05). The mean fractional CSF volume of controls in the four clusters ranged between 0.02 and 0.09 (mean 0.05 ± 0.02), whereas the range in patients was 0.05–0.17 (mean 0.10 ± 0.05); the patients, thus, had twice as much CSF in these cerebellar locations than the controls.
Discussion
In this sample of CJD patients, the neurological syndrome was consistent with expectations [20], showing symptoms referable to cerebral cortex, basal ganglia, brain stem, and cerebellum. The MRI findings were also typical [8, 21] revealing abnormalities, notably hyperintense DWI and FLAIR, in cortex and basal ganglia with a good sensitivity to neurological predictions. However, standard MRI failed to reveal cerebellar or brain stem abnormalities in any patient, despite careful documentation of neurological signs thought to arise from these regions. Because it is possible that patients previously studied by MRI happened to lack cerebellar symptomatology, we specifically selected the current sample for such signs, yet the MRI reading was entirely negative in cerebellum and brain stem, even when using the sequences with highest known sensitivity to CJD, DWI, and FLAIR. This is a topographically specific finding as shown by the good sensitivity of our MRI procedures to cortical and basal ganglia signs.
The cerebellum is prominently affected in CJD. Cerebellar signs are common in patients [22], present in 33% at presentation and in 71% during the course of the disease [11]. The prevalence of cerebellar signs in patients with the E200K familial CJD is 43% [23]. Cerebellar degeneration is commonly found in human CJD neuropathology and in animal models. Loss of granule cells, spongiosis in the granular and molecular layers, and variable involvement of Purkinje cells with occasional formation of axonal torpedoes are the classical abnormalities. Abnormal PrP deposition is evident [7] as well as ultrastructural changes, including reduction of dendritic arbors, loss of dendritic spines, and hypertrophy of dendritic branches of Purkinje cells [24]. Transgenic mice expressing the mutated PrP with an insertion in the octarepeat domain of the prion protein display atrophy of the cerebellum, with reduction in the number of granule cells and a decrease in the thickness of the molecular layer [25]. The cerebellum is also physiologically related to fundamental basal ganglia circuits and is typically activated by deep brain stimulation from the basal ganglia [26], thus confirming direct anatomical connectivity which could be associated with prion propagation. Clinical signs suggesting brainstem involvement are less common and are evident in about 7% of the sporadic CJD patients [22]. Pathological alterations in the brain stem were reported to a variable degree in autopsies of ten consecutive sporadic CJD patients [27]. The quadrigeminal plate and pontine nuclei were the most severely and consistently affected structures, and although spongiform changes were found only infrequently, the immunoreactivity against PrP was seen in all these structures.
Despite its definite involvement in the neurological and pathological phenomenology of CJD, the cerebellum is very rarely noted in imaging studies of CJD. There is some weak evidence of reduced blood flow and reduced 18F-fluoro-2-deoxy-d-glucose uptake in the cerebellum [28] from positron emission tomography (PET) studies, but MRI very rarely produces evidence of tissue abnormality. Poon et al. [29] reported one case with the Brownell–Oppenheimer (cerebellar) variant of CJD with clear DWI (and weaker FLAIR) cerebellar hyperintensity. Lewis et al. [30] reported similar findings in one case of hGH-related iCJD, but two other cases revealed no cerebellar MRI findings, despite clinical cerebellar signs. In fact, this paradoxical pattern of negative cerebellar MRI with proven neuropathological degeneration postmortem is common in the CJD literature [31, 32]. Our patients were examined by trained neurologists that specifically and methodically documented cerebellar symptoms in those patients; yet, MRI, including DWI and FLAIR, was negative in the cerebellum. Thus, standard MRI cannot reveal the cerebellar degeneration of CJD even when it is clearly documented both clinically and neuropathologically.
This finding is specific to the degeneration of CJD. Although FLAIR is known to have lower sensitivity to posterior fossa lesions of some types than to supratentorial lesions [33], it can detect the cerebellar lesions of Wernicke's encephalopathy [34]. DWI appears effective in detecting reduced diffusion in acute posterior fossa stroke [35], and Wernicke's encephalopathy [34], as well as a variety of other neurological conditions including acute stroke [36], inflammatory [37], neoplastic [38], and metabolic [39] disorders. Therefore, it must be that these MRI sequences (FLAIR and DWI) are specifically blind to the histopathological process of CJD in the cerebellum, while being highly sensitive to the changes in cortex and basal ganglia.
In the current experiment, we found cerebellar atrophy, manifested as both elevated CSF volumes by morphometric VBM analysis and elevated diffusion by quantitative ADC diffusion. The atrophy appeared focal and bilateral, at the interface of the cerebellar nodule with the fourth ventricle. This focal atrophy could not be detected on clinical reading of standard MRI sequences but was highly significant on statistical group analyses and quite substantial: CSF volumes in these locations were twice as high in the patients as in the controls and ADC values about 30% higher.
These findings may explain why cerebellar changes are not detected by routine MRI, even in cases where cerebellar involvement is clinically and neuropathologically unambiguous. It appears that degenerative changes are reflected in elevated diffusion in the cerebellum, presumably due to tissue destruction and replacement by CSF, whereas restricted diffusion is the hallmark of the disease in basal ganglia and cerebral cortex. Atrophy of cerebellar tissue would replace some tissue components with fluid or shift water across compartments, causing elevated diffusivity. Such fluid accumulation and loss of tissue may only weakly affect T2 and thus be hard to detect by FLAIR. The DWI images would show reduced, rather than enhanced, signal; because the standard DWI scans are optimized for detection of restricted diffusion, they miss the cerebellar changes. This is the case in another cerebellar syndrome, ataxia telangiectasia, which presents ADC elevations in the absence of detectible FLAIR or DWI findings in children [40]. In animal models of cerebral ischemia, cell lysis and membrane disruption preferentially increase the extracellular volume and allow less restricted movement of water molecules, increasing the apparent diffusion coefficient and leading to pseudonormalization in DWI [41]. In prion diseases, Macfarlane et al. [42] suggested that inherited forms, but not other, display cerebellar atrophy. Our finding of atrophy around the fourth ventricle in mostly genetic E200K patients is similar to the two cases they show, with the 6-OPRI and P102L mutations.
While our current data indicate a solution to the apparent paradox of cerebellar MRI insensitivity, they cannot address the intriguing histological differences between cerebellar and cerebral degenerative changes in CJD. It is assumed that CJD-specific hyperintensities detected by diffusion-weighted MRI in basal ganglia and cortex result from cell damage with characteristic vacuolization of the neuropil [43]. The presence of vacuoles with a diameter of less than 20 μm, gliosis, or astrocytosis restricts the diffusion compared with normal tissue, resulting in an increased DWI signal [44]. Extracellular deposition of abnormal PrP may also contribute to restricted diffusion [45]. These, presumably, are the processes largely responsible for the MRI changes in the cerebrum but not in the cerebellum. There are other curious discrepancies between cerebellar and cerebral expression of CJD neuropathology. Iwasaki et al. [46] reported PrP deposition but no spongiform degeneration or neuronal loss in a patient with autopsy proven CJD and brainstem symptomatology. It is also possible that the cerebellum is affected by a slightly different form of PrPSc, as has been shown in one case [9, 47], or that vacuolization, neuronal loss, and gliosis may appear later in the cerebellum, where the cerebellar Purkinje cells were initially negative for PrPSc [10]. There is also greater white matter involvement reported in the cerebellum [48]. The pathological process in cerebellum of CJD patients may be therefore somehow different from that in the basal ganglia. Nevertheless, when the cerebellum is the site of greatest prion deposition and vacuolation, as has been reported in one case of idiopathic CJD, it also demonstrates reduced diffusion and DWI hyperintensity [31], confirming the reasoning above.
In conclusion, cerebellar degeneration in CJD may be uniquely different from supratentorial changes, at least early in the course of the disease. Strong DWI and FLAIR hyperintensities of the basal ganglia are characteristic to CJD [8, 21] (in the appropriate clinical context). These are associated with reduced ADC and are thought to reflect one or more CJD-specific degenerative aspects, although their histologic determinants are not yet established [44]. In the cortex, both reductions and elevations of ADC exist, confounding detectibility. In the cerebellum, apparently, only elevated diffusion is associated with degeneration, making it undetectable to DWI and FLAIR imaging. This would also explain why PET scanning can possibly detect cerebellar and cortical, but not basal ganglia, abnormalities in CJD [28]. If confirmed by future work, these results suggest that examination of quantitative ADC maps, volumetric analyses, and the detection of cerebellar high diffusivity, could add to the diagnosis and monitoring of CJD.
References
Jakob A (1921) Uber eigenaritge erkrankungen des zentral-nervensystems mit bemerkenswertem anatomischen befunde (spastische pseudosklerose-encephalomyelopathie mit disseminierten degenerationsherden). Z Gesamte Neurol Psychiatr 64:147–228
Hsiao K, Meiner Z, Kahana E, Cass C, Kahana I, Avrahami D et al (1991) Mutation of the prion Protein in Libyan Jews with Creutzfeldt-Jakob disease. N Engl J Med 324:1091–1097
Snowden JS, Mann DM, Neary D (2002) Distinct neuropsychological characteristics in Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry 73:686–694
MacDonald ST, Sutherland K, Ironside JW (1996) Prion protein genotype and pathological Phenotype studies in sporadic Creutzfeldt–Jakob disease. Neuropathol Appl Neurobiol 22:285–292
Brown P, Kenney K, Little B, Ironside J, Will R, Cervenáková L et al (1995) Intracerebral Distribution of infectious amyloid protein in spongiform encephalopathy. Ann Neurol 38:245–253
Masters CL, Richardson EP Jr (1978) Subacute spongiform encephalopathy (Creutzfeldt-Jakob Disease). The nature and progression of spongiform change. Brain 101:333–344
Parchi P, Capellari S, Gambetti P (2000) Intracerebral distribution of the abnormal isoform of the prion protein in sporadic Creutzfeldt-Jakob disease and fatal insomnia. Microsc Res Tech 50:16–25
Young GS, Geschwind MD, Fischbein NJ, Martindale JL, Henry RG, Liu S et al (2005) Diffusionweighted and fluid-attenuated inversion recovery imaging in Creutzfeldt-Jakob disease: high sensitivity and specificity for diagnosis. AJNR Am J Neuroradiol 26:1551–1562
Ferrer I, Puig B, Blanco R, Martí E (2000) Prion protein deposition and abnormal synaptic protein expression in the cerebellum in Creutzfeldt-Jakob disease. Neuroscience 97(4):715–726
Tanaka S, Saito M, Morimatsu M, Ohama E (2000) Immunohistochemical studies of the PrP(CJD) deposition in Creutzfeldt-Jakob disease. Neuropathology 20:124–133
Cooper SA, Murray KL, Heath CA, Will RG, Knight RS (2006) Sporadic Creutzfeldt-Jakob disease with cerebellar ataxia at onset in the UK. J Neurol Neurosurg Psychiatry 77:1273–1275
Kretzschmar HA, Ironside JW, DeArmond SJ, Tateishi J (1996) Diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Arch Neurol 53:913–920
Chapman J, Cohen O, Nitzan Z, Tsabari R, Prohovnik I (2007) The CJD rating scale: a new tool for evaluation of neurologic symptomatology in early CJD patients. Neurology 68:A141
Folstein MF, Folstein SE, McHugh PR (1975) “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12(3):189–198
Stejskal E, Tanner J (1965) Spin diffusion measurements: spin echoes in the presence of a timedependent field gradient. J Chem Phys 42:288–292
Mazziotta J, Toga A, Evans A, Fox P, Lancaster J, Zilles K et al (2001) A probabilistic atlas and reference system for the human brain: International Consortium for Brain Mapping (ICBM). Philos Trans R Soc Lond B Biol Sci 356:1293–1322
Ashburner J, Friston KJ (2005) Unified segmentation. Neuroimage 26:839–851
Ashburner J, Friston KJ (2000) Voxel-based morphometry–the methods. Neuroimage 11:805–821
Rowland L (2005) Signs and symptoms in neurological diagnosis. In: Rowland L (ed) Merritt's textbook of Neurology, 11th edn. Lippincott Williams & Wilkins, Baltimore, pp 60–63
Chapman J, Brown P, Goldfarb LG, Arlazoroff A, Gajdusek DC, Korczyn AD (1993) Clinical heterogeneity and unusual presentations of Creutzfeldt-Jakob disease in Jewish patients with the PRNP codon 200 mutation. J Neurol Neurosurg Psychiatry 56:1109–1112
Fulbright RK, Kingsley PB, Guo X, Hoffmann C, Kahana E, Chapman JC et al (2006) The imaging appearance of Creutzfeldt-Jakob disease caused by the E200K mutation. Magn Reson Imaging 24:1121–1129
Brown P, Gibbs CJ Jr, Rodgers-Johnson P, Asher DM, Sulima MP, Bacote A et al (1994) Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol 35:513–529
Kahana E, Zilber N, Abraham M (1991) Do Creutzfeldt-Jakob disease patients of Jewish Libyan origin has unique clinical features? Neurology 41:1390–1392
Ferrer I (2002) Synaptic pathology and cell death in the cerebellum in Creutzfeldt-Jakob disease. Cerebellum 1:213–222
Chiesa R, Piccardo P, Ghetti B, Harris DA (1998) Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron 21:1339–1351
Phillips MD, Baker KB, Lowe MJ, Tkach JA, Cooper SE, Kopell BH et al (2006) Parkinson disease: pattern of functional MR imaging activation during deep brain stimulation of subthalamic nucleus–initial experience. Radiology 239:209–216
Shintaku M, Yutani C, Doh-ura K (2006) Brain stem lesions in sporadic Creutzfeldt-Jakob disease: a histopathological and immunohistochemical study. Neuropathology 26:43–49
Engler H, Lundberg PO, Ekbom K, Nennesmo I, Nilsson A, Bergström M et al (2003) Multitracer study with positron emission tomography in Creutzfeldt-Jakob disease. Eur J Nucl Med Mol Imaging 30:85–95
Poon MA, Stuckey S, Storey E (2001) MRI evidence of cerebellar and hippocampal involvement in Creutzfeldt-Jakob disease. Neuroradiology 43:746–749
Lewis AM, Yu M, DeArmond SJ, Dillon WP, Miller BL, Geschwind MD (2006) Human growth hormone-related iatrogenic Creutzfeldt-Jakob disease with abnormal imaging. Arch Neurol 63:288–290
Oppenheim C, Zuber M, Galanaud D, Detilleux M, Bolgert F, Mas JL et al (2004) Spectroscopy and serial diffusion MR findings in hGH-Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry 75:1066–1069
Pauri F, Amabile G, Fattapposta F, Pierallini A, Bianco F (2004) Sporadic Creutzfeldt-Jakob disease without dementia at onset: clinical features, laboratory tests and sequential diffusion MRI (in an autopsy-proven case). Neurol Sci 25:234–237
Bastianello S, Bozzao A, Paolillo A, Giugni E, Gasperini C, Koudriavtseva T et al (1997) Fast spin-echo and fast fluid-attenuated inversion-recovery versus conventional spin-echo sequences for MR quantification of multiple sclerosis lesions. AJNR Am J Neuroradiol 18:699–704
Lapergue B, Klein I, Olivot JM, Amarenco P (2006) Diffusion weighted imaging of cerebellar lesions in Wernicke's encephalopathy. J Neuroradiol 33:126–128
Kumral E, Celebisoy M, Celebisoy N, Canbaz DH, Calli C (2007) Dysarthria due to supratentorial and infratentorial ischemic stroke: a diffusion-weighted imaging study. Cerebrovasc Dis 23:331–338
Engelter ST, Wetzel SG, Radue EW, Rausch M, Steck AJ, Lyrer PA (2004) The clinical significance of diffusion-weighted MR imaging in infratentorial strokes. Neurology 62:574–580
Axer H, Ragoschke-Schumm A, Bottcher J, Fitzek C, Witte OW, Isenmann S (2005) Initial DWI and ADC imaging may predict outcome in acute disseminated encephalomyelitis: report of two cases of brain stem encephalitis. J Neurol Neurosurg Psychiatry 76:996–998
Quadery FA, Okamoto K (2003) Diffusion-weighted MRI of haemangioblastomas and other cerebellar tumors. Neuroradiology 45:212–219
Jan W, Zimmerman RA, Wang ZJ, Berry GT, Kaplan PB, Kaye EM (2003) MR diffusion imaging and MR spectroscopy of maple syrup urine disease during acute metabolic decompensation. Neuroradiology 45:393–399
Firat AK, Karakas HM, Firat Y, Yakinci C (2005) Quantitative evaluation of brain involvement in ataxia telangiectasia by diffusion weighted MR imaging. Eur J Radiol 56:192–196
Pierpaoli C, Righini A, Linfante I, Tao-Cheng JH, Alger JR, Di Chiro G (1993) Histopathologic correlates of abnormal water diffusion in cerebral ischemia: diffusion-weighted MR imaging and light and electron microscopic study. Radiology 189:439–448
Macfarlane RG, Wroe SJ, Collinge J, Yousry TA, Jager HR (2007) Neuroimaging findings in human prion disease. J Neurol Neurosurg Psychiatry 78:664–670
Demaerel P, Heiner L, Robberecht W, Sciot R, Wilms G (1999) Diffusion-weighted MRI in sporadic Creutzfeldt-Jakob disease. Neurology 52:205–208
Mittal S, Farmer P, Kalina P, Kingsley PB, Halperin J (2002) Correlation of diffusion-weighted magnetic resonance imaging with neuropathology in Creutzfeldt-Jakob disease. Arch Neurol 59:128–134
Haık S, Dormont D, Faucheux BA, Marsault C, Hauw JJ (2002) Prion protein deposits match magnetic resonance imaging signal abnormalities in Creutzfeldt-Jakob Disease. Ann Neurol 51:797–799
Iwasaki Y, Iijima M, Kimura S, Iwasaki Y, Iijima M, Kimura S et al (2006) Autopsy case of sporadic Creutzfeldt-Jakob disease presenting with signs suggestive of brainstem and spinal cord involvement. Neuropathology 26:550–556
Puoti G, Giaccone G, Rossi G, Canciani B, Bugiani O, Tagliavini F (1999) Sporadic Creutzfeldt-Jakob disease: co-occurrence of different types of PrP(Sc) in the same brain. Neurology 53:2173–2176
Cruz-Sanchez F, Lafuente J, Gertz HJ, Stoltenburg-Didinger G (1987) Spongiform encephalopathy with extensive involvement of white matter. J Neurol Sci 82:81–87
Acknowledgment
The study was supported by NIH grant # NS043488, and parts of these data were previously presented at the 59th Annual Meeting of the American Academy of Neurology, Boston, MA, April 28 to May 5, 2007. We thank Professor Amos Korczyn, Professor Esther Kahana, and Dr. Peter Kingsley for discussions on this paper, and Dr. Sergei Pakhomov, Institute of the Human Brain, Russian Academy of Science, Saint Petersburg, Russia, for the development of MSU.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cohen, O.S., Hoffmann, C., Lee, H. et al. MRI Detection of the Cerebellar Syndrome in Creutzfeldt–Jakob Disease. Cerebellum 8, 373–381 (2009). https://doi.org/10.1007/s12311-009-0106-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12311-009-0106-8