
Abstract
Cerebellar hemorrhage (CH) is a well-known complication in newborns. Among metabolic patients, it has been classically described but rarely reported. This is the first description of a patient with propionic acidemia in whom magnetic resonance imaging (MRI) allowed diagnosis of asymptomatic CH. Due to the usual silent presentation of CH at early ages, we suggest the possibility of including a brain MRI study as part of the routine neurological evaluation in metabolic patients, especially when neurological signs appear.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Cerebellar hemorrhage (CH) in newborns is a well-known complication of traumatic births, extracorporeal membrane oxygenation, coagulation abnormalities, acute septic shock, artero-venous malformations, and disseminated intravascular coagulation [1]. In newborns, CH has been observed in 5–10% of autopsies, and this figure increases to 15–25% in deceased premature infants with fewer than 32 weeks of gestational age and a birth weight under 1,500 g [2, 3].
In metabolic patients, neonatal CH has been considered a classical neurological complication of organic acidemias. However, although it was first described some time ago, to date only three cases have been recorded in the literature. Fischer and colleagues reported one case in a patient with isovaleric acidemia in 1981 [4]. Later, Dave and colleagues described CH in patients with propionic acidemia and methylmalonic acidemia, respectively, in 1984 [5]. The diagnosis was made in one case by neuroimaging (computed tomography—CT) [5]. Conversely, in the others, neonatal CH was an unexpected neuropathologic finding [4, 5]. Moreover, CH was highly suspected of being the cause of death in all of them. Since 1984, no other case of CH in organic acidemias has been reported although many publications with brain CT scan and magnetic resonance imaging (MRI) findings in these metabolic disorders have been published [6–9]. We present a patient with propionic acidemia in whom a silent CH was unexpectedly diagnosed with brain MRI.
Case Report
A 2.7-kg girl was delivered at term following an unremarkable pregnancy and delivery. Second degree consanguinity was recognized by her parents. The Apgar score was 9/10/10 at 1, 5, and 10 min, respectively, and no immediate problems were observed. Two days later, she became lethargic and hypotonic with poor feeding. Blood test revealed metabolic acidosis and hyperammonemia (488 μmol/L). Urine organic acid analysis disclosed an increased concentration of propionate, 3-hydroxypropionate, and methylcitrate, which suggested a diagnosis of propionic acidemia. Propionyl coenzyme A carboxylase activity in cultured fibroblasts was deficient (6.8% of control values), and genetic analysis revealed a homozygous deletion in exon 23 in mRNA of the propionyl coenzyme A carboxylase gene. At this time, pharmacological inotropic support for arterial hypotension, parental nutrition, and veno-venous hemofiltration was started. A normalization of clinical and laboratory parameters was gradually seen by the fourth day and complete enteral feeding was achieved in 12 days. Hemofiltration was extended for 10 days because of oligoanuria due to pre-renal failure secondary to the hemodynamic instability during the acute phase. Progressive jaundice and acholia appeared during the first months of life and progressive signs of cholestasis appeared in blood tests. Prothrombine time decreased to 43% and elevated hepatic enzymes were also found. A complete study to clarify liver failure etiology was performed as this is not a common complication among patients with propionic acidemia. The abdominal ultrasound study revealed mild hepatomegaly, and liver biopsy showed bile duct reduction and diffuse hemochromatosis. Cholestasis was attributed to multifactor etiology related to blood transfusions, hemofiltration, parenteral nutrition, sepsis due to Serratia marcencens, and pharmacological treatment. Parental nutrition length was 15 days, and it was complemented with specialized enteral formula (methionine, threonine, valine, and isoleucine free) and whole protein for 12 days to obtain protein intakes of 130 kcal/kg/day and a maximum of 3 g/kg/day. Despite therapy with ursodesoxicolic acid, the signs of cholestasis and hepatic failure continued.
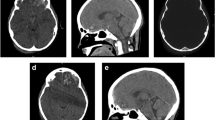
Following this, the patient’s metabolic parameters remained stable with whole protein (0.9 g/kg/day) in addition to the specialized formula (2 g/kg/day), 120 kcal/kg/day, carnitine (100 mg/kg/day), and isoleucine (200 mg/kg/day). Weight and length gain were poor and they remained in the second percentile below the mean for sex and age on the National Center for Health Statistics growth charts. At the age of 2 months, head growth was unsatisfactory, and subsequent brain MRI was performed on a 1.5-T MRI instrument (SignaHD, General Electric, Milwaukee, WI, USA). Sagittal T1, axial T2-weighted FSE, coronal fast fluid-attenuated inversion recovery (FLAIR), axial 3D-FSPGR IR, and diffusion T2-weighted images were obtained. The MR images revealed mild generalized brain atrophy with multiple hypointense millimetric focus in T2 and gradient T2-weighted images localized in cerebellar gray matter. The axial T1-weighted images showed one hyperintense focus in the left cerebellar hemisphere (Fig. 1). The images suggested multiple focus of CH at different stages. No perceptible clinical signs were present. Because of the poor neurological outcome, liver transplant was contraindicated, and the patient died at the age of 6 months due to liver failure secondary to intractable cholestasis.

a, b Axial T2 and gradient T2*-weighted MR images showing multiple hypointense millimetric focus in both hemispheres of the cerebellar gray matter. c Axial T1-weighted image with one millimetric hyperintense signal in the left cerebellar hemisphere
Discussion
Propionic acidemia is a genetic disorder of amino acid metabolism caused by a deficiency of propionyl coenzyme A carboxylase. The clinical presentation may occur at any age, but in most patients, it occurs in the newborn period, precipitated by the initiation of proteins in the diet. The first clinical symptoms are lethargy, poor feeding, vomiting, hypotonia, seizures, and coma. Biochemically, this disorder is characterized by acidosis, hyperammonemia, and moderate lactacidemia [10]. A previous report published in 1994 by Brismar and Ozand [6] described the neuroradiological features of brain CT scan or MRI in 43 patients newly diagnosed with organic acidemias and reviewed 26 cases already reported in the literature. The features found in this report are similar to those of methylmalonic and propionic acidemia, but they seem to be more severe in the latter case. The authors’ findings included white matter abnormalities (in some cases with subcortical white matter necrosis) during the first months of life. Widening of sulci and enlargement of cerebrospinal fluid spaces are usually evidenced at the end of the first year. Additionally, a delay in myelination and lesions affecting the putamen and caudate nucleus are more often detected after the first year of life. In this article, only one case of intracranial bleeding secondary to thrombocytopenia was described but the localization was not specified [6].
CH in organic acidemias may be the result of various factors such as CNS edema due to accumulation of abnormal organic acids, thrombocytopenia (a common laboratory finding), coagulopathy secondary to liver disease, and anticoagulation therapy during veno-venous hemofiltration. These last four situations may have played an important role in our patient, but these conditions are normally associated with extracranial bleeding or hemorrhaging in other areas of the brain (subarachnoid, intra- or periventricular, and white matter diffuse petechial hemorrhage) [5]. There are no bibliographic references in the literature about selective cerebellum involvement in these conditions, although it may be caused by increased sensitivity of this organ to edema and cytotoxicity secondary to the metabolic disorder, with a favorable background provided by the rest of the conditions. Despite the fact that in our patient CH was not clinically evident, this complication turned out to have an important effect on her long-term prognosis. Moreover, it seemed to be a definite factor in the short-term prognosis of the three patients with CH already reported.
In conclusion, CH is probably a misdiagnosed complication in organic acidemias because brain MRI is not part of a routine examination. However, it is probably a very rare complication. In fact, as noted above, only a few cases have been reported, even in long series [6]. Taking into account the fact that CH can remain asymptomatic during the first months of life, a brain MRI study is advisable as part of the routine neurological evaluation of patients with propionic acidemia. Acute neurological signs in the context of normal biochemical exam results should raise the suspicion of CH, and an urgent cranial ultrasound, CT scan, or MRI should be requested. Early ventricular drainage or surgical evacuation does not seem to be useful in the case of small and diffuse hemorrhages, but may change the prognosis in case of extensive bleeding, which probably accounts for a rather small group of patients [11]. In any case, in light of the outcome of our patient and other reported cases, CH in a patient with organic acidemia must be seen as a bad prognostic sign.
References
Merrill JD, Piecuch RE, Fell SC, Barkovich AJ, Goldstein RB (1998) A new pattern of cerebellar hemorrhages in preterm infants. Pediatrics 102:E62
Volpe J (1995) Intracranial hemorrhage: subdural, primary subarachnoid, intracerebellar, intraventricular (term infant) and miscellaneous. In: Volpe J (ed) Neurology of the newborn, 3rd edn. Saunders, Philadelphia, pp 392–393
Perlman JM, Nelson JS, McAlister WH, Volpe JJ (1983) Intracerebellar hemorrhage in a premature newborn: diagnosis by real-time ultrasound and correlation with autopsy findings. Pediatrics 71:159–162
Fischer AQ, Challa VR, Burton BK, McLean WT (1981) Cerebellar hemorrhage complicating isovaleric acidemia: a case report. Neurology 31:746–748
Dave P, Curless RG, Steinman L (1984) Cerebellar hemorrhage complicating methylmalonic and propionic acidemia. Arch Neurol 41:1293–1296
Brismar J, Ozand PT (1994) CT and MR of the brain in disorders of the propionate and methylmalonate metabolism. Am J Neuroradiol 15:1459–1473
Trinh BC, Melhem ER, Barker PB (2001) Multi-slice proton MR spectroscopy and diffusion-weighted imaging in methylmalonic acidemia: report of two cases and review of the literature. Am J Neuroradiol 22:831–833
Andreula CF, De Blasi R, Carella A (1991) CT and MR studies of methylmalonic acidemia. Am J Neuroradiol 12:410–412
Bergman AJ, Van der Knaap MS, Smeitink JA, Duran M, Dorland L, Valk J et al (1996) Magnetic resonance imaging and spectroscopy of the brain in propionic acidemia: clinical and biochemical considerations. Pediatr Res 40:404–409
Fenton WA, Gravel RA, Rosenblatt DS (2001) Disorders of propionate and methylmalonate metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease, 8th edn. McGraw-Hill, New York, pp 2165–2195
Chadduck WM, Duong DH, Kast JM, Donahue DJ (1995) Pediatric cerebellar hemorrhages. Child Nerv Syst 11:579–583
Acknowledgments
Thanks are due to Dr. Celia Pérez-Cerdá and Dr. Magdalena Ugarte of the Centro de Diagnóstico de Enfermedades Moleculares, UAM, Madrid, for enzymatic and genetic studies, and Dr. Antonia Ribes of the Institut de Bioquímica Clínica de Barcelona.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Velasco-Sánchez, D., Gómez-Lopez, L., Vilaseca, M.A. et al. Cerebellar Hemorrhage in a Patient with Propionic Acidemia. Cerebellum 8, 352–354 (2009). https://doi.org/10.1007/s12311-009-0103-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12311-009-0103-y