
Abstract
A recombinant inbred line mapping population of intra-species upland cotton was generated from a cross between the drought-tolerant female parent (AS2) and the susceptible male parent (MCU13). A linkage map was constructed deploying 1,116 GBS-based SNPs and public domain-based 782 SSRs spanning a total genetic distance of 28,083.03 cM with an average chromosomal span length of 1,080.12 cM with inter-marker distance of 10.19 cM.A total of 19 quantitative trait loci (QTLs) were identified in nine chromosomes for field drought tolerance traits. Chromosomes 3 and 8 harbored important drought tolerant QTLs for chlorophyll stability index trait while for relative water content trait, three QTLs on chromosome 8 and one QTL each on chromosome 4, 12 were identified. One QTL on each chromosome 8, 5, and 7, and two QTLs on chromosome 15 linking to proline content were identified. For the nitrate reductase activity trait, two QTLs were identified on chromosome 3 and one on each chromosome 8, 13, and 26. To complement our QTL study, a meta-analysis was conducted along with the public domain database and resulted in a consensus map for chromosome 8. Under field drought stress, chromosome 8 harbored a drought tolerance QTL hotspot with two in-house QTLs for chlorophyll stability index (qCSI01, qCSI02) and three public domain QTLs (qLP.FDT_1, qLP.FDT_2, qCC.ST_3). Identified QTL hotspot on chromosome 8 could play a crucial role in exploring abiotic stress-associated genes/alleles for drought trait improvement.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Upland cotton (Gossypium hirsutum L.), is considered a “cash crop”, due to its global demand for natural textile fiber and also a major source of vegetable oil. Yield in upland cotton is highly dependent on irrigation, specifically during the flowering and boll developmental stages (Han 2001). However, the ever-changing climatic condition, especially, water scarcity during the reproductive phase, has drastically reduced cotton production. The effect of drought at a molecular, biochemical, physiological, and morphological level generally varied among different plants (Hasan et al. 2018) but in cotton, water scarcity adversely affects overall plant growth and reduces the number and quality of bolls, thereby leading to reduced fiber yield (Batista et al. 2013). Therefore, the genetic improvement for water stress tolerance in cotton has long been tried but met with limited success (Roy et al. 2011; Schuster 2011; Abdelraheem et al. 2015; Ali et al. 2017).
Drought tolerance traits such as root architecture, cell membrane stability, and relative water content have been studied in-depth to introgress such traits in the elite upland cotton cultivars in breeding programs. However, the lack of a deep understanding of the molecular and physiological aspects of these traits and their effective incorporation limits their progress. Under drought stress, plants undergo several physiological and biochemical changes. Among several parameters, plant height (PH), chlorophyll stability index (CSI), percentage of relative water content (RWC), proline content (PC), chlorophyll content (CC), and nitrate reductase activity (NRA) are the most crucial and emphasized in several studies(Jones et al. 1980; Kumar and Singh 1998; Mohan et al. 2000; Ranawake et al. 2011; Arve et al. 2011; Parkash and Singh 2020; Iqbal et al. 2020).Generally, faster growth leads to taller plants, which eventually require more water. But in water scarcity, growth is likely to be affected severely (Ranawake et al. 2011), and the yield will be substantial. Drought stress leads to osmotic stress and loss of turgor pressure in plants (Arve et al. 2011). It results in plant height and root length reduction, as well as loss of chlorophyll content resulting in yellowing leaves. Limited water supply also results in decreased entry of essential minerals (nitrate, phosphorus, calcium, and potassium) into the plant (Iqbal et al. 2020) for their growth. Drought tolerant genotypes with higher chlorophyll stability are more capable of withstanding drought stress. With higher CSI, genotype results in more photosynthate that helps in higher biomass (Mohan et al. 2000). RWC helps in maintaining turgidity and water levels in cells (Kumar and Singh 1998), thereby drought-tolerant genotypes with higher CSI have more photosynthate that helps in having higher biomass. High RWC in genotypes is a well-known indicator of drought tolerance. A lower level of RWC leads to low photosynthate due to stomatal closure and decreased cell turgidity (Parkash and Singh 2020). Thus, both CSI and RWC are key parameters for drought tolerance evaluation (Kumar and Singh 1998; Mohan et al. 2000). Higher proline content in leaves upon desiccation was recorded as a common response in many crops or plant species (Kannan 2017). Jones et al. 1980 suggest that high proline content helps in osmotic regulation. It prevents hydration of proteins in dried tissues, acts as a storage compound for nitrogen and carbon, thus helps in cellular functions. Nitrogen (N) is equally important as water for plant growth and productivity. Under drought conditions, transport, and uptake of N in above-ground plant parts decrease drastically due to the unavailability of water for uptake (Iqbal et al. 2020) and reduces plant metabolism.
For a couple of decades, functional genomics and DNA markers have thrown light on molecular mechanisms of drought tolerance in plants (Nguyen 1999; Poland 2000; Qin et al. 2009). DNA markers, especially Simple Sequence Repeats (SSRs) and Single Nucleotide Polymorphism (SNPs) have proved their potentials in the genetic dissection of fiber quality under water stress (Baytar et al. 2018). In addition, four drought tolerance-associated candidate genes (RD2, HAT22, PIP2, and PP2C) have been identified by combining GWAS and RNA-seq data in upland cotton (Hou et al. 2018). In another study, late embryogenesis abundant (LEA) proteins have been shown to have a crucial role in protecting cotton under drought stress (Magwanga et al. 2018). Tian et al. (2019)showed that an endo-reduplication-related gene, GaTOP6B used to promote the growth of leaf and roots for controlling drought tolerance in cotton. Drought tolerant trait-linked specific genes, Quantitative Trait Loci (QTL) or QTL clusters can be incorporated into the elite cultivars using a marker-assisted selection strategy. However, such effort in cotton as a sound breeding strategy for drought tolerance is very few in comparison to rice, maize, and sorghum. This may be due to low coverage of the cotton genome with the molecular markers, diversified drought-resistant component traits involved in cotton, and lack of validation of identified genomic segments or QTLs, across the various genetic backgrounds.
The present study was undertaken to construct a genetic map for the intra-specific cross of G. hirsutum and identify drought tolerance QTLs. The resulted genetic map and QTLs in our study were merged with the drought tolerant QTLs of the recent studies, available in the public domain to develop a consensus map to identify QTL cluster-associated with drought tolerance traits.
Material and methods
Plant materials
The two contrasting parents of upland cotton were selected after exposure to drought stress (no irrigation) for 15 days continuously to the full-grown vegetative plants. The intra-specific mapping population of upland cotton was developed by crossing two contrasting parents with drought tolerance trait. The two parents were selected based on the earlier report by Ananthi and Vijayaraghavan (2012). AS2, one upland cotton variety with a higher value of drought characteristics such as RWC, CSI, PC was used as the female drought-tolerant parent while MCU13 which is drought susceptible upland cotton variety with lower value of drought characteristics such as RWC, CSI, and proline content was selected as the male parent.
The F1was grown at the CSIR-NBRI cotton experimental field campus in the year 2015 and forwarded to F2 in 2016. With the single seed descent method, we advanced these populations to F3 and F4 generation in the years 2017 and 2018, respectively. Finally, seeds from the F4 generation were collected and sown in Odisha at a farmer’s cotton field as an F5 generation trial with 122 individuals. The F5 generation plants were exposed to similar drought stress as parents. However, due to reduced viability and cyclone devastation, only 48 individuals of the F5 generation were left for our experimentation. Thus, a total of 48 individuals from the F5 generation were further used for the construction of a genetic map and QTL analysis in the present study.
GBS library preparation, genotyping, and SNP calling
Fresh leaf was used for genomic DNA isolation by a modified CTAB method. Genomic DNA was digested with ApeKI, and the GBS library was constructed. The library was sequenced using the Ion Torrent platform. GBS analysis and SNP calling were performed using the Tassel 5 GBSv2 pipeline (Glaubitz et al. 2014). Raw data FASTQ files were prepossessed using FastQC (Andrews 2010) and Trimmomatic (Bolger et al. 2014) to ensure that data is of good quality without any adapter contamination (Bio-project Accession Number PRJNA638773, Bio-sample Accession Number SAMN15207880 to SAMN15207929). Barcoded sequence reads were collapsed into a set of unique sequence tags and then master tags. The master tags were then aligned to the reference genome of Gossypium hirsutum cultivar TM-1 (Assembly: ASM98774v1, Seq accession: GCF_000987745.1) to form tags on a physical map that depicted the genomic position of each tag. The barcode in the original sequence reads was then used counts in each sample in the tags by taxa (TBT). Eventually, the recorded information in tags on the physical map, tags by taxa were used to call SNPs at each tag locus and filter the SNPs among 48 individuals of the F5 generation. Only those SNPs, which resulted in all 48 individuals of F5with polymorphic alleles, were taken into account and others were rejected in further analysis.
SSR markers and SSR-typing
For polymorphic SSRs, the parental lines were surveyed using ~ 3,000 SSR primer pairs. The SSR primer pair details were obtained from cottongen (https://www.cottongen.org/) with the following options: BNL primers, JESPR primers, TM primers, NBRI primers. A cost-effective method of fluorescent-labeled SSR typing was followed for multiplex SSR PCR (Schuelke 2000). In this method, SSR PCR fragments were fluorescent dye-labeled with a sequence-specific forward primer and reversed primer one-tube PCR reaction. This method allowed high-throughput genetic analyses to improve SSR typing efficiency. The 829 parental polymorphic SSRs were used to screen the RIL mapping population (F5). The chi-square test for goodness of fit was assessed for the Mendelian 1:1 inheritance ratio.
Genetic map construction
The genetic map was constructed by employing polymorphic SSR and SNP markers using ‘IciMapping 4.1’. The feature Kosambi mapping function (Kosambi 1944) in the software was used to convert the recombination frequency into the genetic map distance (CentiMorgan, cM). Linkage groups (LGs) were determined with a logarithm of odds (LOD) score threshold of 3. RECORD algorithm was used for the correct ordering of SNPs across chromosomes. Rippling was also performed for fine-tuning the order of markers by the sum of adjacent recombination frequencies (SARF) with a window size of 5.
Assignment of LGs and collinearity
The developed LGs in the above analysis were assigned to chromosomes using SNPs with known physical positions. The physical location of SNPs was obtained by mapping the sequencing reads on the reference genome Gossypium hirsutum cultivar TM-1 (ASM98774v1).
Analysis of morphological and biochemical traits for drought tolerance
The growth performance and a few biochemical analyses of recombinant inbred lines (F5) in addition to the parental lines were assessed for drought tolerance in terms of plant height (PH), chlorophyll stability index (CSI), percentage of relative water content (RWC), proline content (PC), chlorophyll content (CC), and nitrate reductase activity (NRA). The youngest fully expanded leaves were taken for recording relative water content. Barrs and Weatherley’s (1962) method was adopted for RWC% and it was expressed in percentage.
Similarly, CSI was calculated by the method described by Murty and Majumder (1962). By the method of Bates et al. (1973), the PC was estimated and expressed in μg g−1fresh weight. Similarly, by adopting the method of Nicholas and Shah (1976), the NRA was determined and expressed as μg of NO2−g−1 h−1. CC was measured according to the method described by Arnon and Whatley 1949 using the following formula:
The trait distribution was assessed for all physiological and phenotypic traits using the Kolmogorov–Smirnov test.
QTL analysis
QTL analysis was performed using QTL IciMapping 4.1 software with the Inclusive composite interval mapping (ICIM) method. The identified QTLs were located for drought tolerance under a single environment. No multi-environmental conditions were considered for the QTL analysis. ICIM was performed with a window of 0.2 cM probability in stepwise regression of 0.001. For significant QTLs of each trait, either LOD threshold of ≥ 2.5 or R2 value of ≥ 0.5 was considered with 1,000 permutation test at P = 0.05. The comparative QTL clustering was made taking all the drought tolerance QTLs and genetic maps from the latest field drought-related study by Abdelraheem et al. (2018) into the context.
Construction of consensus map and combined QTL ANALYSIS
A consensus map was constructed with IciMappingV4.2 (Meng et al. 2015). “CMP” tool was used to make the consensus map by integrating our in-house high-density genetic linkage map and one of the public domain linkage maps with SNP/SSRs markers and drought trait QTLs. The algorithm used was “nnTwoOpt”, the linkage was calculated using the “SAD” option, and grouping was done as a single group with a window size of 5. QTL clustering was performed for our in-house data and public domain data (Abdelraheem et al. 2018). The information file of consensus mapping reveals only chromosome 8 had the common markers to be carried out for the QTL clustering for the drought-associated QTLs. All the identified QTLs in the present study and public domain study were analyzed for QTL clustering.
Results
GBS sequencing and its data analysis
Ion Torrent sequencing generated 319,598,638 reads (~ 47 GB), which were processed and cleaned for removing the adaptors and contaminants. A total of 293,808,976 (~ 41.9 GB) cleaned reads were obtained and subjected to further analyses (Supplemental data S1). In these cleaned reads, short sequence sample barcode analysis was performed and resulted in 160,438,503 (54.61%) sequencing reads with sample barcodes for further downstream analysis of separate tag counts and master tag counts. The separate tag counts were merged into a total of 96,063,846master tag counts (Supplemental data S1). Only master tags occurring at/or above 48 minimum number of reads across all of the FASTQ files in the experiment were retained in the output master tag list as good master tags. Finally, good master tags were mapped against the reference genome G. hirsutum cultivar TM-1 (ASM98774v1). Overall, 60.15% (635,202/1,055,954) of good master tags were able to map with the reference genome, and 39.85% (420,752/1,055,954) of good master tags remained unmapped (Supplemental data S1). Comparing the above tags information on physical map of cotton (ASM98774v1, GCF_000987745.1), a total of 108,272 SNP sites were identified (Table 1). However, two parental lines of the above-mentioned 48 RILs population resulted in 41,702 polymorphic SNP sites with sequencing data of ~ 240 MB in AS2 and ~ 197 MB in MCU13. When the same polymorphic SNP calls of parental lines were compared with the SNP calls of 48 RILs population, it resulted in 2,834 common polymorphic SNPs. There are many SNP calls specific to each RILs that could not be included further.
Genetic map construction and consensus mapping
A total of 3,663 polymorphic markers (829 SSRs and 2,834 SNPs) between both parents were initially included for the genetic map construction. After filtering > 20% binning SNPs (i.e. 1,262 SNPs), and 503 excluded markers (including both SSR and SNP markers), a total of 1,898 markers (1,116 SNP and 782 SSRs) were finally mapped across 26 chromosomes (Fig. 1) and formed a genetic map (Supplemental data S2). The genetic map harbored a total distance of 28,083.03 cM with an average inter-marker distance of 10.19 cM (Table 2). Large numbers of SNPs were mapped on chromosomes 5, 15, 19, 20 and 21. Among SSRs, > 50 SSRs were allocated on chromosomes 5 and 26while 35 SSRs each on chromosomes 2 and 11. Chromosome 5 was found to be the largest chromosome with a maximum number of 126 markers and a span length of 1,732.13 cM. Chromosome 23 was found to be the shortest chromosome with an overall span length of 442.83 cM and 24 mapped markers. A total of 923 loci were mapped in At sub-genome (A01-A13) with a total span length of 14,432.45 cM with an average marker distance of 10.9 cM. Whereas, Dt sub-genome (D01-D13) consisted of 975 markers with a full span length of 13,650.58 cM and an average marker distance of 9.4 cM. We constructed the consensus map taking both the maps of in-house as well as public domain by Abdelraheem et al. (2018). Six SSR markers were found to be common in both maps. A maximum of three SSRs was found to be common in chromosome 8 in the consensus map (Fig. 2a).



Genetic map showing molecular linkage groups of the intra-species cross of small RIL population for AS2 and MCU13 and QTLs of field drought traits. Map distances between adjacent markers are represented in centimorgan (cM), Various QTLs represented in the right side of map with designed block and name

Consensus map of chromosome 8 and QTL cluster analysis taking QTL and map data of in-house mapping population from the cross AS2 x MCU13 and public domain data of drought tolerance traits (Abdelraheem et al. (2018); a QTL clustering on consensus map of Chromosome 8, b Enlarged view of QTL clustering on chromosome 8
Evaluation of the physiological and biochemical parameters
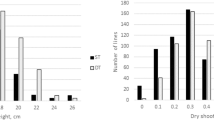
Plant height (PH) revealed significant differences between the two parental lines as well as in the Recombinant Inbred Lines (RILs) (Fig. 3). The AS2 was estimated to a higher PH than the other parent, MCU13 (the average PH of AS2 is 35.3 cm and the average PH of MCU13 is 31. 7 cm). The mean PH of RILs of AS2xMCU13 was 44.9 cm. A gradual increase in plant height was also noticed throughout the growth period of RILs, however water deficit significantly reduced the plant height than the parents. Similarly, data on total chlorophyll content (TCC) recorded at the vegetative stage significantly influenced over parents. Higher total chlorophyll content of 1.0075 mg/g of AS2 was observed than 0.8975 mg/g of MCU13. The genotype AS2 maintained higher total chlorophyll content in all stages than others even under the stress conditions. The total chlorophyll values were significantly lower in the case of MCU13. The mean value of total chlorophyll content of the RIL population was1.025 mg/g while the variance was 0.012 mg/g at the vegetative stage. The statistical test of the normal distribution of TCC data was also represented in Table 3. The proline content (PC) was revealed to increase when drought stress is subjected to the plant. There was also an increase in PC concerning the age of the plant, however, it was found to be increased till the flowering stage and decreased till the boll development stage. A mean quantity of 848.00 μgg-1 of proline was recorded at the vegetative stage of AS2 while other parent, MCU13 showed a decrease in the PC (823.25 μgg-1) without irrigation. Contrary to the parental lines, RILs under drought stress showed a mean PC value of 816.29 μgg-1. The details of the distribution test are represented in Table 3. As mean data on nitrate reductase activity (NRA) was recorded at different stages, a higher value of NRA was observed with AS2 (9.16 μg of NO2 g−1 h−1) in contrast to MCU13 (8.55 μg of NO2 g−1 h−1). The overall NRA estimate varied significantly among RILs with a range from 7.92 to 8.86 μg of NO2 g−1 h−1. Between two parental lines, AS2 recorded a higher value of relative water content (RWC) percentage than MCU13. However, the RILs of AS2x MCU13 recorded a mean of 77.30 RWC percentage.

Box plot analysis of RILs in addition to the parental lines for physiological and biochemical traits related to drought tolerance; Plant height (PH), Chlorophyll stability index (CSI), Percentage of relative water content (RWC), Proline content (PC), Chlorophyll content (CC), and Nitrate Reductase Activity (NRA)
QTL analysis and drought QTL clustering
QTL clustering was performed on a consensus genetic map and a total of 19 QTLs were identified in nine different chromosomes (chromosome 3, 4, 5, 7, 8, 12, 13, 15, and 26) (Supplemental data S3, S4). Chromosomes 3 and 8 harbored important drought tolerance QTLs for CSI traits. The qtlCSI01 was harboured on chromosome 3 and spanned by the SNP markers of NBRI_250675 and NBRI_27009576. The other three QTLs (qtlCSI02, qtlCSI03, and qtlCSI04) were contained on chromosome 8. Three QTLs of RWC (qtlRWC01, qtlRWC03, qtlRWC05) were identified on chromosome 8 and two QTLs of RWC (qtlRWC02 and qtlRWC04) were identified on chromosome 4 and chromosome 12, respectively. There were five QTLs identified for each biochemical drought trait such as PC and NRA. The qtlPC01, qtlPC02, and qtlPC03 were found on chromosome 8, 5 and 7, respectively while chromosome 15 contained two PC QTLs (qtlPC04, qtlPC05). Similarly, two QTLs of NRA (qtlNRA02, qtlNRA03) were harboured on chromosome 3 while the other three QTLs of NRA such as qtlNRA01, qtlNRA04, qtlNRA05 were contained on chromosomes 8, 13, and 26. Besides the above-mentioned QTLs, one important QTL (i.e. qtlTCC01) was found on chromosome 8 spanning in between NBRI_e0509 and NBRI_30441289. All the details of the QTLs are represented in supplemental data S3, S4 for their respective LOD, position, flanking SNPs, r2 value, and α significance value (Table 4). The QTL clustering analysis on chromosome 8 revealed that the two QTLs of CSI (qCSI01, qCSI02) were clustered with three QTLs (qLP.FDT_1, qLP.FDT_2, and qCC.ST_3) of the public domain data of Abdelraheem et al. (2018) resulting in drought tolerance QTL hotspot on chromosome 8 (Fig. 2a, b). It was further explored that in addition to this QTL hotspot, there was another QTL cluster of six drought QTLs of the public domain within a range of nearly 3 cM (Fig. 2b,). We tried to explore the uniqueness of the QTLs clustered on chromosome 8, however synteny analysis revealed that chromosome 8 shared similarity with other chromosomes of G. hirsutum too.
Discussion
Drought is undoubtedly one of the major constraints to all crop production leading to a substantial decline in yields (Dabbert and Gore 2014). The restricted plant growth and development is the result of drought stress by altering metabolic activity and biological function (Fahad et al. 2015) of the plant evolved with several cellular and molecular make-ups to overcome the drought. Drought tolerance is a complex and multi-factor trait involving several signalling pathways and various molecular responses through differentially expressed genes (Price et al. 2002). QTLs mapping is one of the appropriate approaches to dissect the genetic basis of drought tolerance (Ashraf 2010). However, the high degree of interaction between QTL and the environment is the major flaw for which the selection of an appropriate QTL for drought tolerance is hindered (Tuberosa and Salvi 2006). Thus, once a QTL is identified for drought tolerance, its characterization with structural organization and validation is indeed a vital role for crop improvement programs (Salvi and Tuberosa 2005). Genetic maps are considered for rapid progress towards the improvement of crops for complex traits like drought tolerance. With the recent developments and technologies in plant sciences, a detailed in-depth understanding of drought tolerance is still underway.
GBS sequencing and sequencing data
Genotyping by Sequencing (GBS) is one of the powerful tools in molecular breeding and has the potential to bridge the genotypic data with specific traits. More specifically, GBS lowers the cost of molecular work of marker-genotyping through the recent advancement of NGS technology. Thus, GBS is the single approach that is generally used to produce a large amount of marker data in a cost-effective way, for which many researchers prefer the GBS approach for high-density genetic mapping for QTL analysis.
High-density genetic mapping
In this study, a high-density map has been developed with a total of 1,898 markers including 1,116 SNPs and 782 SSRs. Thus, the high-density map of both marker systems (SSRs and SNPs) resulted from this study will have wider applications and suit for the consensus mapping. There are a dozen genetic maps in the public cotton domain, however, it will be unique among many genetic maps harboring both types of markers. This map can comply with earlier reported SSR-based as well as recent development of SNP-based genetic maps. One genetic map based on cotton SNP80K assay with 12,116 SNPs (Tan et al. 2018), two genetic maps based on 63 K SNParray containing 22,829 SNPs (Hulse-Kemp et al. 2015), and genetic map based on IntlCottonSNPConsortium_70K Chip with 2,393 SNPs (Zhang et al. 2017) were comparable with our present map. In addition, the genetic linkage map is the basis of all QTL analysis and dissects the genetic basis of important traits. It also plays a fundamental role in understanding the structure of the genome and explains various events of evolution. In contrast to the above-cited SNP-based linkage maps, Yu et al. (2012) developed a linkage map with 2,072 loci (1,825 SSRs and 247 SNPs) like our genetic map. However, the number of SSRs and SNPs were reverse in our developed genetic linkage map. This is only possible due to the recent advancement of high throughput DNA sequencing technology like RNA sequencing, gene-enriched sequencing, or sequencing of reduced representational libraries. Unlike our developed genetic map, where 26 LGs were assigned, many other genetic maps could not resolve/assign all the loci into 26 LGs due to various reasons. For example-Wang, et al. (2015a, b) used 1,013 loci and they were able to map all these loci into 50 LGs with 41 linkage groups assigned to 23 chromosomes. Cotton researchers also used to have the obvious advantages of SNPs, InDels, ESTs and then used these important genomic resources to develop the genetic linkage map like Li et al. (2014) in which 1,349 EST-based SNPand InDel markers were screened and the genetic map could be possible only with 137 loci. This showed another bottleneck of cotton genetic mapping, where low polymorphism is a huddle to overcome on the way towards high-density genetic mapping. In such context, the present developed genetic map in our study has advantages, with a large number of SNPs like other available public domain genetic maps. Besides, a few hundred SSRs can also play an additional advantage for consensus mapping as the unique characteristic of co-linearity.
The genetic maps are generally constructed using different types and sizes of mapping populations. All these sizes and types of mapping population can affect the efficiency of mapping because of the differences in the genetic distance between markers and the degree of recombination in various crossovers during division. The size of the experimental mapping population thus exerts an influence on the accuracy of genetic maps. The higher number of individuals in the mapping population, the more precise is the map. However, 200 individuals were considered being sufficient for the construction of a reasonably accurate genetic map (He et al. 2001). In the present study, the mapping population was small, which was unintentional due to natural calamity as mentioned in the material and methods section. As studied mapping population is RILs, unlike BC and F2 populations, it would not get hugely affected in the genetic distances because of minimal recombination frequency errors. The same was discussed by He et al. (2001) in their work. Further, it is still inconclusive to determine the ideal number of individuals of a mapping population for establishing an accurate genetic map (Ferreira et al. 2006). With above context, our genetic map is relatively dense with lesser inter marker genetic distance and less prone to genetic distance errors. The span length of our genetic map was found to be larger than usual. However, for such mapping span length expansion, there are many factors such as the size and type of mapping population (Lee et al. 2015; Bai et al. 2016) and few other factors such as segregation distortion, excess of heterozygosity, genotyping errors, missing values, and mapping software (N’Diaye et al. 2017). Similar map expansion has also been observed previously in various crops such as potato (Tanksley et al. 1992), tomato (Paran et al. 1995), and maize (Anderson et al. 2003). However, the high map span length due to the small mapping population size in the present study could not be ignored.
Segregation distortion is widespread in mapping populations. It is relatively serious in RILs because of genetic drift (Zhang et al. 2009) linked with natural and artificial selection for several generations (Xian-Liang et al. 2006). In the present study, the frequency of segregation distortion was relatively higher than the previously reported map. This suggests that RIL development may probably contribute to more distorted segregation. The same conclusion was also found in several reports on cotton (Wang et al. 2015; Li et al. 2016; Zhang et al. 2016; Zhang et al. 2017). The earlier reported RILs-based SNP maps for upland cotton exhibited segregation distortion ranging from 13.29 to 63.77% (Wang et al. 2015; Li et al. 2016; Zhang et al. 2016; Zhang et al. 2017). This infers that the divergence level of the parental lines of the mapping population may also play a critical role in segregation distortion (Paterson et al. 1988).
Physiological and biochemical parameters
The distribution of PH data and NRA of the mapping population was out of range of that of the parents. This may resulted due to raising of parents and mapping populations in different climatic zone. Under drought conditions, nitrate accumulates mostly in the roots and least in the leaves; Thus, NRA activity got reduced in our drought experiment. The drought tolerant parent, AS2 showed higher NRA activity while in relatively drought sensitive parent, MSU13, NRA activity was in the range of mapping population. Hence the difference of NRA activity from the parents is fairly expected. The RWC of the mapping population was not out of the parental range rather it is more towards AS2 parental type (drought tolerant). CSI index of the mapping population was also in the parental range. It is more towards MCU13 type (drought susceptible). The proline and chlorophyll content of the mapping population was also in the parental range. Thus, all the physiological and biochemical data was not out of range of parental type except plant height and nitrate reductase activity.
Consensus mapping and drought QTL cluster linked to physiological and biochemical analysis
In the present study, 19 QTLs related to drought stress characteristics were identified in cotton on a high-density intra-specific genetic map on nine different chromosomes such as chromosome 3, 4, 5, 7, 8, 12, 13, 15, and 26 under single environmental condition. In the same line, many cotton researchers reported QTLs for drought traits. Abdelraheem et al. (2018) like our study reported a total of 150 QTLs for field drought tolerance and 80 QTLs for salt tolerance in green-house conditions. Similarly, Zheng et al. (2016) studied 11 morphological and physiological traits under water-limited (W1) and well-watered (W2) regimes at three growth stages (bud, flowering, and full boll) and reported four consistent QTLs under water-limited condition while no consistent QTLs were found in well-watered condition. In our study, QTL analysis has not been performed under multi-environmental conditions, for which we could not infer the Q X E effect with stable/consistent QTL. As we consider only one environment in our study for the identification of QTLs simulating drought stress, it may not be majorly affected by other minor environmental factors. The identified QTLs are standalone for the studied environment and they may be influenced slightly with trait value when those are studied in other environments.
The small size mapping population followed the “Beavis effect” in general for which there is a possibility of the inaccuracy of QTL position. However, the inter-maker distance in our study was relatively small (10.90 cm in the At-subgenome and 9.47 cM in the Dt-subgenome) for which QTL position will be reliable and accurate. Besides, we performed composite interval mapping (CIM) for QTL identification that may not lead to any false positive like SMA analysis. The same effect of small mapping population sizes was also discussed on the reliability of QTL mapping (Raghavan and Collard 2012). Most of the identified QTLs are definite and bear reliable information through stringent criteria with LOD of ≥ 2.5 or R2 value of ≥ 0.5. In our study, out of 19 QTLs chromosome 8 harbored the maximum number of identified QTLs (Three QTLs for CSI trait, three QTLs for RWC trait, one QTL for PC, three QTLs for NRA trait, and one important QTL for TCC) with comparison to other chromosomes. Due to a lack of common markers on chromosomes between two maps (one in-house developed map with SNPs and SSRs and the other public domain reported map) in other chromosomes, a consensus map for chromosome 8 was only possible. A consensus map of chromosome 8 revealed an island having drought tolerance QTL hotspot with two QTLs of CSI (qCSI01, qCSI02) from the present study and three QTLs (qLP.FDT_1, qLP.FDT_2, and qCC.ST_3) of the public domain data of Abdelraheem et al. (2018). Similar to our finding on chromosome 8, Zheng et al. (2016) reported earlier a consistent QTL on chromosome 8 for field drought tolerance with few biochemical and physiological characteristics.
The drought QTL cluster on chromosome 8 from our study will be a novel genomic resource. It could be further characterized to identify few essential genes involved in governing the drought tolerance in cotton. In a similar context, Abdelraheem et al. (2017) also performed a QTL cluster analysis for abiotic stress in cotton which helped in the identification of a candidate gene and opened the gateway for molecular breeding programs. Thus, it is assumed that the present identified drought tolerance QTL hotspot can be characterized in Indian cotton varieties for a better understanding of the molecular and genetic mechanism underlying abiotic stress tolerance.
Conclusion
In this study, a genetic linkage map containing 1,116 SNPs and 782 SSRs was constructed, which spanned 28,083.03 cM of span length with an average distance of 10.19 cM between markers. Nineteen QTLs were identified in nine chromosomes (chromosome-3, 4, 5, 7,8,12, 13, 15 and 26) for field drought tolerance traits such as plant height (PH), chlorophyll stability index (CSI), relative water content (RWC), proline content (PC), chlorophyll content (CC), nitrate reductase activity (NRA). Further, one QTL hotspots on chromosome 8 inferred from the consensus-based mapping approach with the mapping data of public domain (Abdelraheem et al. 2018). All these findings were credible and deserved priority for fine mapping to identify candidate genes/alleles of drought tolerance in upland cotton and further be deployed in marker-assisted selection (MAS) in cotton breeding programs.
References
Abdelraheem A, Fang DD, Zhang J (2018) Quantitative trait locus mapping of drought and salt tolerance in an introgressed recombinant inbred line population of Upland cotton under the greenhouse and field conditions. Euphytica 214:1–20
Abdelraheem A, Liu F, Song M, Zhang JF (2017) A meta-analysis of quantitative trait loci for abiotic and biotic stress resistance in tetraploid cotton. Mol Genet Genomics 292:1221–1235
Abdelraheem A, Mahdy E, Zhang J (2015) The first linkage map for a recombinant inbred line population in cotton (Gossypium barbadense) and its use in studies of PEG-induced dehydration tolerance. Euphytica 205:941–958
Ali F, Bano A, Fazal A (2017) Recent methods of drought stress tolerance in plants. Plant Growth Regul 82:363–375
Ananthi K, Vijayaraghavan H (2012) Rapid determination of soluble protein content, nitrate reductase activity and yield studies in cotton genotypes under water stress. Int J Food Agri Vet Sci 2(1):147–152
Anderson LK et al (2003) High-resolution crossover maps for each bivalent of Zea mays using recombination nodules. Genetics 165:849–865
Andrews S (2010) FastQC: a quality control tool for high throughput sequence data [Online]. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Arnon DI, Whatley F (1949) Is chloride a coenzyme of photosynthesis? Science 110:554–556
Arve LE, Torre S, Olsen JE, Tanino KK (2011) Stomatal responses to drought stress and air humidity. In: Shanker A, Venkateswarlu B (eds) Abiotic stress in plants - mechanisms and adaptations. IntechOpen. https://doi.org/10.5772/24661 Available online at: https://www.intechopen.com/chapters/18406
Ashraf M (2010) Inducing drought tolerance in plants: recent advances. Biotechnol Adv 28:169–183
Bai Z-Y, Han X-K, Liu X-J, Li Q-Q, Li J-L (2016) Construction of a high-density genetic map and QTL mapping for pearl quality-related traits in hyriopsis cumingii. Sci Rep 6:1–11
Barrs H, Weatherley P (1962) A re-examination of the relative turgidity technique for estimating water deficits in leaves. Aust J Biol Sci 15:413–428
Bates LS, Waldren RP, Teare I (1973) Rapid determination of free proline for water-stress studies. Plant Soil 39:205–207
Batista CH, de Aquino LA, Silva TR, Silva HRF (2013) Crescimento e produtividade da cultura do algodão em resposta a aplicação de fósforo e métodos de irrigação. REVISTA BRASILEIRA DE AGRICULTURA IRRIGADA-RBAI 4:197
Baytar AA, Peynircioğlu C, Sezener V, Basal H, Frary A, Frary A, Doğanlar S (2018) Identification of stable QTLs for fiber quality and plant structure in Upland cotton (G. hirsutum L.) under drought stress. Ind Crops Prod 124:776–786
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120
Dabbert T, Gore MA (2014) Challenges and perspectives on improving heat and drought stress resilience in cotton. J Cotton Sci 18:393–409
Fahad S, Hussain S, Matloob A, Khan FA, Khaliq A, Saud S, Hassan S, Shan D, Khan F, Ullah N (2015) Phytohormones and plant responses to salinity stress: a review. Plant Growth Regul 75:391–404
Ferreira A, da Silva MF, Cruz CD (2006) Estimating the effects of population size and type on the accuracy of genetic maps. Genet Mol Biol 29:187–192
Glaubitz JC, Casstevens TM, Lu F, Harriman J, Elshire RJ, Sun Q, Buckler ES (2014) TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PloS one 9:e90346
Han H (2001) Experiment and study on effects of moisture coerce on cotton production. Trans CSAE 17:37–40
Hasan M, Ma F, Prodhan Z, Li F, Shen H, Chen Y, Wang X (2018) Molecular and physio-biochemical characterization of cotton species for assessing drought stress tolerance. Int J Mol Sci 19:2636
He P, Li J, Zheng X, Shen L, Lu C, Chen Y, Zhu L (2001) Comparison of molecular linkage maps and agronomic trait loci between DH and RIL populations derived from the same rice cross. Crop Sci 41:1240–1246
Hou S, Zhu G, Li Y, Li W, Fu J, Niu E, Li L, Zhang D, Guo W (2018) Genome-wide association studies reveal genetic variation and candidate genes of drought stress related traits in cotton Gossypium hirsutum L.). Front Plant Sci 9:1276
Hulse-Kemp AM, Lemm J, Plieske J, Ashrafi H, Buyyarapu R, Fang DD, Frelichowski J, Giband M, Hague S, Hinze LL (2015) Development of a 63K SNP array for cotton and high-density mapping of intraspecific and interspecific populations of Gossypium spp. G3: Genes Genomes, Genetics 5:1187–1209
Iqbal A, Dong Q, Wang X, Gui H, Zhang H, Zhang X, Song M (2020) High nitrogen enhance drought tolerance in cotton through antioxidant enzymatic activities, nitrogen metabolism and osmotic adjustment. Plants 9:178
Jones M, Osmond C, Turner N (1980) Accumulation of solutes in leaves of sorghum and sunflower in response to water deficits. Funct Plant Biol 7:193–205
Kannan V, Srinivasan G, Babu R, Thiyageswari S, Sivakumar T (2017) Response of relative leaf water content, chlorophyll stability index, proline, and yield of cotton to the application of biochar, mulch and ppfm spray under differing moisture regimes. Int J Curr Microbiol App Sci 6:604–611
Kosambi D (1944) The estimation of map distance. Ann Eugenics 12:505–525
Kumar A, Singh D (1998) Use of physiological indices as a screening technique for drought tolerance in oilseedbrassicaspecies. Ann Bot 81:413–420
Lee M et al (2015) A consensus linkage map of oil palm and a major QTL for stem height. Sci Rep 5:1–7
Li C, Dong Y, Zhao T, Li L, Li C, Yu E et al (2016) Genome-wide SNP linkage mapping and QTL analysis for fiber quality and yield traits in the upland cotton recombinant inbred lines population. Front Plant Sci 7:1356. https://doi.org/10.3389/fpls.2016.01356
Li X, Gao W, Guo H, Zhang X, Fang DD, Lin Z (2014) Development of EST-based SNP and InDel markers and their utilization in tetraploid cotton genetic mapping. BMC Genomics 15:1–11
Mohan M, Narayanan SL, Ibrahim S (2000) Chlorophyll stability index (CSI): its impact on salt tolerance in rice. Int Rice Res Notes 25:38–39
Magwanga RO, Lu P, Kirungu JN, Lu H, Wang X, Cai X, Zhou Z, Zhang Z, Salih H, Wang K (2018) Characterization of the late embryogenesis abundant (LEA) proteins family and their role in drought stress tolerance in upland cotton. BMC Genet 19:6
Meng L, Li H, Zhang L, Wang J (2015) QTL IciMapping: integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J 3:269–283
Murthy KS, Majumdar SK (1962) Modifications of the technique for determination of chlorophyll stability index in relation to studies of drought resistance in rice. Curr Sci 31:470–471
N’Diaye A, Haile JK, Fowler DB, Ammar K, Pozniak CJ (2017) Effect of co-segregating markers on high-density genetic maps and prediction of map expansion using machine learning algorithms. Front Plant Sci 8:1434
Nguyen H (1999) Molecular dissection of drought resistance in crop plants: from traits to genes. In: Ribaut JM, Poland D (eds) Molecular approaches for the genetic improvement of cereals for stable production in water-limited environments. Mexico D.F, CIMMYT, pp 36–40
Nicholas D, Shah Y (1976) Carbon monoxide oxidation over a platinum-porous fiber glass supported catalyst. Ind Eng Chem Prod Res Dev 15:35–40
Paran I, Goldman I, Tanksley S, Zamir D (1995) Recombinant inbred lines for genetic mapping in tomato. Theor Appl Genet 90:542–548
Parkash V, Singh S (2020) A review on potential plant-based water stress indicators for vegetable crops. Sustainability 12:3945
Paterson AH, Lander ES, Hewitt JD, Peterson S, Lincoln SE, Tanksley SD (1988) Resolution of quantitative traits into Mendelian factors by using a complete linkage map of restriction fragment length polymorphisms. Nature 335:721–726. https://doi.org/10.1038/335721a0
Ribaut J-M, Poland D (eds) (2000) Molecular approaches for the genetic improvement of cereals for stable production in water-limited environments. A strategic planning workshop held at CIMMYT, El Batan, Mexico, 21–25 June 1999. Mexico DF, CIMMYT
Price AH, Cairns JE, Horton P, Jones HG, Griffiths H (2002) Linking drought-resistance mechanisms to drought avoidance in upland rice using a QTL approach: progress and new opportunities to integrate stomatal and mesophyll responses. J Exp Bot 53:989–1004
Qin Y, Liu R, Mei H, Zhang T, Guo W (2009) QTL mapping for yield traits in Upland cotton (Gossypium hirsutum L.). Acta Agron Sin 35:1812–1821
Raghavan C, Collard B (2012) Effect of small mapping population sizes on reliability of quantitative trait locus (QTL) mapping. Afr J Biotechnol 11:10661–10674
Ranawake A, Dahanayaka N, Amarasingha U, Rodrigo W, Rodrigo U (2011) Effect of water stress on growth and yield of mung bean (Vigna radiata L). Tropical Agric Res Ext 14:76–79
Roy SJ, Tucker EJ, Tester M (2011) Genetic analysis of abiotic stress tolerance in crops. Curr Opin Plant Biol 14:232–239
Salvi S, Tuberosa R (2005) To clone or not to clone plant QTLs: present and future challenges. Trends Plant Sci 10:297–304
Schuster I (2011) Marker-assisted selection for quantitative traits. Crop Breed Appl Biotech 11:50–55
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotech 18:233–234
Tan Z, Zhang Z, Sun X, Li Q, Sun Y, Yang P, Wang W, Liu X, Chen C, Liu D (2018) Genetic map construction and fiber quality QTL mapping using the CottonSNP80K array in upland cotton. Front Plant Sci 9:225
Tanksley S et al (1992) High density molecular linkage maps of the tomato and potato genomes. Genetics 132:1141–1160
Tian Y, Gu H, Fan Z, Shi G, Yuan J, Wei F, Yang Y, Tian B, Cao G, Huang J (2019) Role of a cotton endoreduplication-related gene, GaTOP6B, in response to drought stress. Planta 249:1119–1132
Tuberosa R, Salvi S (2006) Genomics-based approaches to improve drought tolerance of crops. Trends Plant Sci 11:405–412
Wang H, Jin X, Zhang B, Shen C, Lin Z (2015a) Enrichment of an intraspecific genetic map of upland cotton by developing markers using parental RAD sequencing. DNA Res 22:147–160
Wang Y, Ning Z, Hu Y, Chen J, Zhao R, Chen H et al (2015b) Molecular mapping of restriction-site associated DNA markers in allotetraploid Upland Cotton. PLoS ONE 10:e0124781. https://doi.org/10.1371/journal.pone.0124781
Xian-Liang S, Xue-Zhen S, Tian-Zhen Z (2006) Segregation distortion and its effect on genetic mapping in plants. Chinese J Agri Biotech 3:163–169
Yu JZ et al (2012) A high-density simple sequence repeat and single nucleotide polymorphism genetic map of the tetraploid cotton genome. G3 Genes Genomes Genetics 2:43–58. https://doi.org/10.1534/g3.111.001552
Zheng J et al (2016) Mapping QTLs for drought tolerance in an F2: 3 population from an inter-specific cross between Gossypium tomentosum and Gossypium hirsutum genetics and molecular research. GMR. https://doi.org/10.4238/gmr.15038477
Zhang Z, Shang H, Shi Y, Huang L, Li J, Ge Q et al (2016) Construction of a high-density genetic map by specific locus amplified fragment sequencing (SLAF-seq) and its application to quantitative Trait Loci (QTL) analysis for boll weight in upland cotton (Gossypium hirsutum.). BMC Plant Biol. https://doi.org/10.1186/s12870-016-0741-4
Zhang Z, Ge Q, Liu A, Li J, Gong J, Shang H, Shi Y, Chen T, Wang Y, Palanga KK (2017) Construction of a high-density genetic map and its application to QTL identification for fiber strength in upland cotton. Crop Sci 57:774–788
Zhang Z-S et al (2009) Construction of a comprehensive PCR-based marker linkage map and QTL mapping for fiber quality traits in upland cotton (Gossypium hirsutum L.). Mol Breed 24:49–61
Acknowledgements
We would like to thank Dr. Soon Joo Yap, Codon Genomics S/B, JalanDutamas 7, 43200 Seri Kembangan, Selangor, Malaysia for his kind help in data analysis and valuable suggestions. We acknowledge further Dr. A.B. Das, Utkal University and Dr. P.K. Singh to help us in improving this MS.
Funding
Director, CSIR-National Botanical Research Institute.
Author information
Authors and Affiliations
Contributions
SNJ conceptualized the idea while RPS took it in his Ph.D. research work. All the experiments were done by RPS under the supervision of SNJ. However, SBK helped in SNP typing analysis, and GJT analyzed the genetic mapping discussing with SNJ. RPS drafted the MS with the help of GJT. SNJ, NMB, BJ, and GJT rigorously edited and revised the whole manuscript. The authors do not have any conflict of interest during the entire process of conducting the experiments, data analysis, and preparation of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Authors declare no conflict of interest.
Ethical approval
Institutional Committee (CSIR-NBRI_MS/2020/06/12).
Consent to participate
All the authors have given their consent to participate in the submitted manuscript.
Consent for publication
All the authors have given their consent for publication of the submitted manuscript in physiology and molecular biology of plants.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Shukla, R.P., Tiwari, G.J., Joshi, B. et al. GBS-SNP and SSR based genetic mapping and QTL analysis for drought tolerance in upland cotton. Physiol Mol Biol Plants 27, 1731–1745 (2021). https://doi.org/10.1007/s12298-021-01041-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12298-021-01041-y