
Abstract
Breast cancer is the most prominent cause of cancer death in women worldwide. The highlights of this review are to provide an overview of the targeted therapeutic agents, challenges with metastatic breast cancer (MBCa), mechanisms of action through Hedgehog/Gli 1 signaling pathway and future prospective. Over a decade of success, several drugs have been approved and are in the advanced stages of clinical trials that target the receptors such as estrogen receptor, growth factor receptor, receptor activator of nuclear factor kappa-B, etc. Currently, several monoclonal antibodies are also used for the treatment of breast cancer. Advances in understanding tumor biology, particularly signaling pathways such as Notch signaling pathway, Hedgehog/Gli 1 signaling pathway, and inhibitors are considered to be important for bone metastasis. These studies may provide vital information for the design and development of new strategies with respect to efficacy, reduction of the side effects, and treatment strategies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cancer is one of the most dreaded diseases worldwide. Unfortunately, enhancements in socioeconomic circumstances are associated with increasing cancer incidences such as breast, blood, lung, oral, prostate cancer, etc. [1, 2]. Of these, breast cancer (BCa) is one of the most common cancers endangering women [3, 4], constituting 14.6 % of all cancers [5]. National Cancer Institute (NCI) has estimated that the diagnosis of 246,660 new cases and 40,450 deaths due to BCa in the United States in 2016 and still the incidence is rising [5]. There are several types of BCa, such as ductal carcinoma in situ, invasive ductal carcinoma, triple negative breast cancer (TNBC), inflammatory breast cancer, MBCa, and other types. The efficacy of developing therapeutic options for BCa patients is time-limited and non-curative. These treatments are provided by a single agent or in combination that dependent on disease stages, histology and molecular subtypes, and menopausal status [6]. These treatments have numerous side effects and often unsuccessful to remove the tumor completely. Hence, to overcome these drawbacks, perpetual screening for extremely safer drugs has been ongoing numerous decades, resulting in the finding of new anticancer drugs and vaccines significantly.
Estrogen receptor as pharmacological target
The relation between hormones and malignancy growth has been recognized more than a century. Estrogens play a major role in promoting the proliferation both the normal and neoplastic breast epithelium [7]. Estrogen receptor (ER) is a member of the nuclear hormone family of intracellular receptors translocating into the nucleus and bind to DNA then regulate the activity of different genes [8]. ERα and ERβ are two different forms of ER produced from two separate genes that are differentially expressed in the tissue [9]. ERs are over-expressed in around 70 % of breast cancer cases, referred to as ER-positive (ER+), and can be demonstrated in such tissues using immunohistochemistry. In the mammary gland, estradiol (E2) binds to ERα and ERβ that control cell proliferation and differentiation [10]. They are coded by two distinct genes located on chromosomes 6 and 14 that produce two proteins with 595 and 530 amino acids, respectively [11]. The ER comprises of five main regions that include the N-terminal domain (NTD), a conserved DNA-binding domain (DBD), variable hinge region, a conserved ligand binding domain (LBD) and variable C-terminal region with diverse roles in signaling [12]. ER+ breast cancers are estrogen-dependent and include luminal types A and B. ER-negative
(ER−) breast cancers are estrogen independent and include subtypes in which human epidermal growth factor receptor 2 (HER2) also known as ErbB2 is over-expressed
Current treatment options
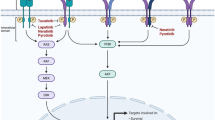
Various types of BCa treatment are currently available to treat pre- and postmenopausal women including surgery, brachytherapy, endocrine therapy, chemotherapy, vaccine treatment and targeted therapy, bone directed treatment, etc. Of these, Surgery (mastectomy) and radiotherapy play an important role in the treatment of BCa in earlier stages. Systemic therapy may be used for almost all BCa patients predominantly for those with advanced stage [13]. Hormone receptor-positive cancers are often treated with hormone-blocking therapy. Aromatase inhibitors (AIs) are the preferred option for postmenopausal women. The monoclonal antibodies or other immune-modulators may be administered in certain cases of MBCa. Several therapeutic targets and signaling pathways are explained in Fig. 1.

Schematic diagram of the therapeutic targets and signaling pathways of the active drugs
Notch signaling pathway
Notch signaling pathway is a potential therapeutic target for the treatment of BCa that involves in the cell proliferation, differentiation, and apoptosis [14]. There are different types of Notch receptors and ligands that have different effects on the development of tissues and organs. This pathway is one of the more sensitive targets to inhibit BCa stem cell subset, which is resistant to standard treatments such as chemotherapy and radiation [15]. Even if Notch inhibitors alone do not yield major responses and cures, there is growing evidence that synergy can result from combining Notch inhibition with already-existing treatment modalities such as chemotherapy, radiation, and other pathway inhibitors. The first Notch pathway inhibitors used both experimentally and clinically were the γ-secretase inhibitors which prevent cleavage of NEXT and therefore the release of NCID from the plasma membrane [15]. The γ-secretase inhibitors, SAHM1, and TR4 have the benefit that can disrupt signaling by all four Notch receptors [16]. In addition, there are several approaches specifically GSIs, dnMAML1, and inhibitory antibodies available to block Notch signaling. While γ-secretase inhibitors are already in clinical trials as Notch-inhibiting agents and are clinically promising, they are highly nonspecific. Other experimental means of Notch inhibition include γ-secretase inhibitors, peptide or antibody blockers, stapled peptides, and genetic strategies such as RNA interference [17]. The detailed structural information of the drugs targeting Notch pathway is given in Table 1.
Hedgehog/gli1 signaling pathway
The hedgehog (Hh) signaling pathway plays an essential role in the regulation of embryonic development and tissue homeostasis of diverse adult tissues, and its deregulation has been implicated in the tumorigenesis and metastasis of various malignancies including BCa. This pathway is a highly coordinated and orchestrates the network involving in the inhibition of twelve transmembrane protein, Pathed1 (Ptch1) by binding Hh protein, activation of the seven-transmembrane protein, Smoothened (SMO), release of a five-zinc finger transcription factor, Gli from a large protein complex, nuclear translocation of Gli, and transcription of target genes [18]. In the absence of Sonic Hh (Shh), Glisorganize a large protein complex with Costal2, fused and suppressor fused (SUFU) and are sequestered in the cytoplasm [18]. In the presence of Shh, a full-length Gli3 released from the large protein complex is transported into the nucleus to activate Hh target genes [19]. Gli1 is one of target genes of Gli3. Therefore, Gli1 is a marker of the Hh pathway activation [20]. Recently, Targeting the Hh pathway has been recognized to be one of the promising therapies for BCa.
Cyclopamine is the first Hh inhibitor, identified from naturally occurring plant alkaloids [21, 22]. It has poor oral bioavailability and suboptimal pharmacokinetics and thus more potent derivatives have been synthesized. This led to the development of Hh modulators with improved potency and druggability, such asvismodegib (GDC-0449), IPI-926, sonidegib (LDE-225), BMS-833923, PF-04449913, and LY2940680 [22]. The agents inhibiting the Hh are also being tested in women with advanced breast cancer in early clinical trials. Several synthetic, small-molecule SMO antagonists have been developed with higher potency than cyclopamine such as SANT1-SANT4, CUR-61414, HhAntag-691, and GDC-0449 which have been tested in preclinical models against a variety of solid tumors [23].
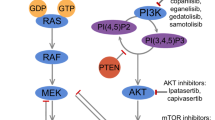
Recently, the in vitro anticancer activities of GANT-61 was reported at the dose ranges from 5 to 20 µM, which significantly reduced the survival of 7/8 tested cell lines after 48 h and in all cell lines after 72 h of treatment relative to the vehicle control [21]. GDC-0449 significantly decreased cell survival in all cell lines only at the highest dose (12 µM) after 72, 96 or 6 days of treatment [21]. Cyclopamine and CUR0199691 have been used successfully in vivo to treat Hh network-induced cancers [22]. Recent in vitro cell line studies provide more insight into the association of Hh signaling with breast cancer. Gli-1 mRNA level was increased in a number of breast cancer cell lines, including MDA-MB-453 (TN), MDA-MB-231 (TN and basal type B), BT20 (basal type A), MCF10A (benign breast cancer cell line), and SKBR3 (HER2+) in comparison to a primary human mammary epithelial cells (HMEC) [19]. RU-SKI 43 is as selective small molecule inhibitor of Hedgehog acyltransferase (Hhat) recently identified and also reduced the growth of ER+ cell proliferation, whereas a structurally related, inactive compound had no effect. Overexpression of Hhat in ER+ cells not only rescued the growth defect in the presence of RU-SKI 43 but also resulted in increased cell proliferation in the absence of drug [24]. Furthermore, the depletion or inhibition of Hhat reduced proliferation of HER2 amplified as well as tamoxifen-resistant cells. Inhibition of Smoothened had no effect on proliferation, indicating that canonical Shh signaling was not operative. If this pathway continues to be of interest, targeting the Hh signaling would be successful in breast cancers (Fig. 2) [24]. The structure, chemical composition, and properties of Hh targeting drugs are given in Table 1.

Schematic diagram of the Hedgehog/gli1 signaling pathways
Drugs targeting bone metastasis and environment
Strategies to target bone metastases have included bisphosphonate therapy, receptor activator of nuclear factor kappa-B ligand (RANK-L) directed monoclonal antibody (mAbs) therapy, and palliative radiation in addition to systemic therapy [25]. The recent preclinical study showed that radium-223 alone or in combination with doxorubicin or zoledronic acid increased survival and reduced serum bone biomarkers in a mouse model of BCa bone metastasis [26]. Cathepsin K is a lysosomal cysteine protease highly expressed in osteoclasts, plays a major role in bone resorption. Odanacatib is a promising compound under phase II clinical investigation in women with BCa and bone metastases [27]. Cilengitide is an RGD-mimetic cyclic pentapeptide inhibitor of both αvβ3 and αvβ5 integrins, inhibits bone metastasis in animals and tested in patients [28]. Saracatinib, a dual inhibitor of Src/Abl, has been shown to decrease levels of bone resorption markers in a phase I study in patients with solid tumors [29]. Dasatinib is a potent, orally available inhibitor by blocking the activity against of multiple oncogenic novel tyrosine kinase inhibitors (TKIs) such as the Src/Ab1 family kinases, a non-receptor or receptor tyrosine kinase that has been recently implicated in MBCa to the bone [30]. The structure, chemical composition, and properties of above mentioned drugs are given in Table 2.
Immunotherapy and immunomodulators
Modern anticancer therapy involves the use of mAbs which is once administered to the patient will selectively target a particular protein involved in the proliferation of tumor cells [31]. A synthetic sialyl-Tn (STn) antigen was generated for use as a therapeutic cancer vaccine antigen, and tests in the animal model and human studies showed the antigen to be safe and to produce a strong immune response [32]. Nivolumab is a fully human IgG4 programmed cell death 1 (PD-1) immune checkpoint inhibitor antibody. It boosts the body’s immune system by targeting a protein on white blood cells called PD-1 [33]. It is currently under investigation in phase II study in randomized, non-comparative trial of Nivolumab after induction treatment in TNBC patients have been started (NCT02499367) [33]. Ipilimumab is an anti-cytotoxic T-lymphocyte antigen 4 (CTLA-4) mAb. Pembrolizumab is a human antibody used in cancer immunotherapy. It targets the programmed cell death 1 (PD-1) receptor [34]. The structures, properties, and function of immunomodulators are given in Table 3.
Inhibition of angiogenesis
Angiogenesis has a key role to play in tumor growth and progression [35]. It switches the shift of the balance between proangiogenic and antiangiogenic in favor of proangiogenesis, applies to all types of solid tumors [36]. Tumor cells enhance local vascular endothelial growth factor (VEGF) production to stimulate the outgrowth of new blood vessels; moreover elevated vascular endothelial growth factor receptor (VEGFR) levels are associated with cancer progression and poor survival rates. Several approaches are employed to inhibit the VEGF pathway. Bevacizumab is a mAb that inhibits VEGFR signaling through attaching and counteracting VEGF-A. It increases response rate (RR) and progression-free survival (PFS) of patients with MBCa when added to first-line chemotherapy in three randomized phase III trials [37]. Ramucirumab is a fully human immunoglobulin G1 mAb (IgG1) developed for the treatment of solid tumors [38]. Sorafenib is an oral multikinase inhibitor that targets Raf-1, wild-type B-Raf, tyrosine kinases acting on VEGFR, platelet-derived growth factor receptor (PDGFR), Flt-3, and c-Kit [39]. Sunitinib malate (SM) is an inhibitor of receptor tyrosine kinases (RTKs) that include VEGFR, PDGFR, stem cell factor receptor (KIT), and colony stimulating factor-1 receptor (CSF1R) [40]. Pazopanib is an oral small molecule TKI of VEGFR, PDGFR, and KIT [41]. Lucitanib is a potent, oral inhibitor of the tyrosine kinase activity of FGFR, PDGFR, and VEGFR [42]. Regorafenib, masitinib, and imatinib mesylate may prove to be clinically useful in inhibiting breast cancer cell migration and metastasis [42]. Axitinib is an oral, potent, selective TKI of VEGFR1, VEGFR2, and VEGFR3 with antiangiogenic and antitumor properties [43]. Aflibercept is an inhibitor of VEGFR. It is a recombinant, decoy receptor fusion protein, rationally designed to block angiogenesis by targeting VEGF-A, VEGF-B, and placental growth factor [44]. The clinically available drugs against angiogenesis are given in Table 4.
Growth factor receptor inhibitors
Growth factors bind to activate RTKs on the high affinity cell surface, which triggers the intracellular signaling pathways [45]. Palbociclib is an oral, small-molecule inhibitor of cyclin-dependent kinases (CDKs) 4 and 6 with preclinical evidence of growth-inhibitory activity in ER+ breast cancer cells and synergy with anti-estrogens [46]. Ado-trastuzumab emtansine and pertuzumab have been FDA-approved for HER2-positive breast cancer [47]. Currently, EGFR (ERB1) and ErbB2 (HER2) oral dual TKI lapatinib reduces tyrosine phosphorylation, as well as activation of extracellular signal-regulated kinase 1/2-mitogen-activated protein kinase (MAPK) and PI3K/AKT, affecting downstream effectors of both proliferation and survival [48]. Afatinib is a novel, orally bioavailable, anilinoquinazoline compound. This agent acts as a potent, irreversible, highly selective inhibitor of EGFR/HER1, HER2, and HER4 tyrosine kinase activity [49]. Gefitinib is an EGFR TKI. Neratinib is an oral irreversible inhibitor of endothelial growth factor receptor and HER2 tyrosine kinases [50]. Panitumumab is a fully human mAb specific to the EGFR [51]. Cetuximab binds to EGFR and prevents its intracellular signaling [52]. Figitumumab, a fully human IgG2α–IGF-IR mAb, generated enthusiasm in a randomized phase II clinical study [53]. Imatinib mesylate (IM) inhibits several protein tyrosine kinases, including PDGFR and c-kit, which are preferentially expressed in tumor cells [42]. MEDI-573, amAb with high binding affinity for both IGFs selectively inhibits the activation of both the IGF1R and IR-A signaling pathways and in mouse models without disrupting glucose metabolism mediated by insulin and IR interaction [54]. AVE1642, a humanized IgG1 antibody, showed promising data in preclinical studies but failed in its phase II clinical trials in breast cancer patients [55]. Ganitumab, a fully humanized IgG1 antibody, is being tested in combination with cytotoxic chemotherapy, mTOR inhibitors, and hormonal therapies in various diseases, including NSCLC, colorectal, pancreatic, ovarian, and breast cancer [56]. Dalotuzumab, another humanized IgG1 antibody with promising preliminary profiles [57], is currently being studied with AIs and the mTOR antagonist in advanced breast cancer. Ruxolitinib inhibits dysregulated Janus kinase (JAK) signaling associated with myelofibrosis. A randomized, double-blind, study of ruxolitinib or placebo in combination with capecitabine in subjects with advanced or metastatic HER2-negative breast cancer is in phase II clinical trial (NCT02120417). Temsirolimus, an inhibitor of mTOR, has clinical activity as intravenous (IV) monotherapy in heavily pretreated locally advanced breast cancer or MBCa [58]. The structures, chemical composition, and properties of the GFR inhibitors are given in Table 5.
Selective estrogen receptor downregulators (SERDs)
SERD is a novel class of compound that modulates the level and activity of the ER and displays the tissue selective activation of estrogenic signaling. It improves the normal function and bone strength without affecting the breast and endocrine system outcomes. These ligands should behave as a potent antagonist in the ER+ cell proliferation with no agonist effects. SERD provides treatment options in a variety of diseases, including resistance to endocrine therapies for ER+ BCa in postmenopausal women [59]. Fulvestrant is an SERD that competitively binds to ER, with a binding affinity approximately 100 times greater than that of tamoxifen [60]. In preclinical studies, fulvestrant has been shown to inhibit the in vitro growth of human breast carcinoma cells and was also effective in tamoxifen-resistant breast carcinoma xenografts in in vivo mouse models. GDC-0810 or ARN-810 is currently in phase II clinical trials. It is an orally active and robust activity in models of tamoxifen-sensitive and tamoxifen-resistant BCa with locally advanced or metastatic ER+ BCa [61]. AZD 9496 is a small molecule that can antagonist activity of ERα and induces receptor degradation in the MCF-7 Xenograft model. Recently, SS5020 has been reported to downregulate the ER with antitumor effects against chemically induced memory tumors [62]. A closely related analog of SS1020 was also reported to be a selective ER down-regulator by the same group. At present, SERD such as SR16234, ZK191703, ZD164384, RU58668, RAD1901, GDC0810 or ARN-810, TAS-108 showed potent bioavailability and good activity in clinical trials (Table 6).
Natural antiestrogen
Thymoquinone (TQ) is isolated from Nigella Sativa that inhibits the growth of cancer cell and induces apoptosis in both ER− and ER+ cancer cell lines [63]. Tehranolide is a natural sesquiterpene lactone isolated from Artemisia annua. It inhibits cell proliferation by affecting PI3K/AKT/cyclin D signaling [63]. Deoxybouvardin is a natural cyclopeptide derived from Rubia Yunnanensis that exhibits a variety of biological activities, including anti-neoplastic. Subglutinol A is an immunosuppressive α-pyrone diterpenoid that can be isolated from Fusarium Subglutinans, an endophytic fungus. It has been proven to be an ER antagonist [64]. The natural effects of I3C are attributable to DIM, which shows evidence of anti-tumorigenic activities in vivo and in vitro by reducing the growth of breast cancer cells [65].
Conclusion
In summary, there are several types of drugs having different pharmacological properties, administration route, and targeting site that have already marketed for the treatment of breast cancer. Many therapeutic options and their mode of activation have been elaborately discussed herewith. The discovery of new targets and expression in breast cancer has been developed and reported by the researchers all over the world, but also further development is ongoing to reduce or minimize the riskiness or side effects. Therefore, further investigation will be important to focus on the design of drugs and new agents acting on targets for validating the concept of the underlying single or combined treatment strategy. Ultimately, the comparative studies of carrier-linked prodrugs with current chemotherapeutic regimens will also be needed for their market approval. This review is to focus on the availability of multiple agents with different ways of mechanisms of activation possessing the challenges and may also be useful for further innovation.
References
Bhandari PR. Crocus sativus L. (saffron) for cancer chemoprevention: a mini review. JTCM. 2015;5:81–7.
Elancheran R, Maruthanila VL, Ramanathan M, et al. Recent discoveries and developments of androgen receptor based therapy for prostate cancer. Med Chem Commun. 2015;6:746–68.
Chen J, Duan Y, Zhang X, et al. Genistein induces apoptosis by the inactivation of the IGF-1R/p-Akt signaling pathway in MCF-7 human breast cancer cells. Food Funct. 2015;6:995–1000.
Carvalho AF, Hyphantis T, Sales PMG, et al. Major depressive disorder in breast cancer: a critical systematic review of pharmacological and psychotherapeutic clinical trials. Cancer Treat Rev. 2014;40:349–55.
Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975–2013, National Cancer Institute. Bethesda. MD, http://seer.cancer.gov/csr/1975_2013/. Based on November 2015 SEER data submission, posted to the SEER web site, April 2016.
Di Leo A, Curigliano G, Dieras V, et al. New approaches for improving outcomes in breast cancer in Europe. Breast. 2015;24:321–30.
Beatson GT. On the treatment of inoperable cases of carcinoma of the mamma: suggestions for a new method of treatment with illustrative cases. Lancet. 1896;2:104–7.
Kumar R, Zakharov MN, Khan SH, et al. The dynamic structure of the estrogen receptor. J Amino Acids. doi:10.4061/2011/812540.
Roy SS, Vadlamudi RK. Role of estrogen receptor signaling in breast cancer metastasis. Int J Breast Cancer. 2012;. doi:10.1155/2012/654698.
Platet N, Cathiard A, Gleizes M, et al. Estrogen and their receptors in breast cancer progression: a dual role in cancer proliferation and invasion. Crit Rev Oncol Hematol. 2004;51:55–67.
Nagaraj G, Ma C. Revisiting the estrogen receptor pathway and its role in endocrine therapy for postmenopausal women with estrogen receptor-positive metastatic breast cancer. Breast Cancer Res Treat. 2015;150(2):231–42.
Kampa M, Pelekanou V, Notas G, et al. The estrogen receptor: two or more molecules, multiple variants, diverse localizations, signaling and functions. Are we undergoing a paradigm-shift as regards their significance in breast cancer? Hormones. 2013;12(1):69–85.
Goldhirsch A, Wood WC, Coates AS, et al. Strategies for subtypes dealing with the diversity of breast cancer: highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer. Ann Oncol. 2011;22:1736–47.
Lee SM, Moon J, Redman BG, et al. A phase II study of RO4929097 gamma-secretase inhibitor in metastatic melanoma: SWOG 0933. Cancer. 2015;121(3):432–40.
Yuan X, Wu H, Xu H, et al. Notch signaling: an emerging therapeutic target for cancer treatment. Cancer Lett. 2015. doi:10.1016/j.canlet.2015.07.048.
Al-Hussaini H, Subramanyam D, Reedijk M, et al. notch signaling pathway as a therapeutic target in breast cancer. Mol Cancer Ther. 2011;10(1):9–15.
Li L, Zhang J, Xiong N, et al. Notch-1 signaling activates NF-jB in human breast carcinoma MDA-MB-231 cells via PP2A-dependent AKT pathway. Med Oncol. 2016;33:1–11.
Ramaswamy B, Lu Y, Teng KY, et al. Hedgehog signaling is a novel therapeutic target in tamoxifen-resistant breast cancer aberrantly activated by PI3K/AKT pathway. Cancer Res. 2012;72(19):5048–59.
Kubo M, Nakamura M, Tasaki A, et al. Hedgehog signaling pathway is a new therapeutic target for patients with breast cancer. Cancer Res. 2004;64:6071–4.
Souzaki M, Kubo M, Kai M, et al. Hedgehog signaling pathway mediates the progression of non-invasive breast cancer to invasive breast cancer. Cancer Sci. 2011;102:373–81.
Kwon YJ, Hurst DR, Steg AD, et al. Gli1 enhances migration and invasion via upregulation of MMP-11 and promotes metastasis in ER alpha negative breast cancer cell lines. ClinExp Metastasis. 2011;28:437–49.
Benvenuto M, Masuelli L, Smaele ED, et al. In vitro and in vivo inhibition of breast cancer cell growth by targeting the Hedgehog/GLI pathway with SMO (GDC-0449) or GLI (GANT-61) inhibitors. Oncotarget. 2016;7:9250–70.
Gonnissen A, Isebaert S, Haustermans K. Targeting the Hedgehog signaling pathway in cancer: beyond Smoothened. Oncotarget. 2015;6:13899–913.
Li YH, Gao HF, Wang Y, et al. Overexpression of Gli1 in cancer interstitial tissues predicts early relapse after radical operation of breast cancer. Chin J Cancer Res. 2012;24(4):263–74.
Wang X, Yang KH, Wanyan P, et al. Comparison of the efficacy and safety of denosumab versus bisphosphonates in breast cancer and bone metastases treatment: a meta–analysis of randomized controlled trials. Oncol Lett. 2014;7:1997–2002.
Coleman R, Aksnes AK, Naume B, et al. A phase IIa, nonrandomized study of radium-223 dichloride in advanced breast cancer patients with bone-dominant disease. Breast Canc Res Treat. 2014;145:411–8.
Clézardin P. Therapeutic targets for bone metastases in breast cancer. Breast Cancer Res. 2011;13:207.
Bäuerle T, Komljenovic D, Merz M, et al. Cilengitide inhibits progression of experimental breast cancer bone metastases as imaged noninvasively using VCT, MRI and DCE-MRI in a longitudinal in vivo study. Int J Cancer. 2011;128(10):2453–62.
Baselga J, Cervantes A, Martinelli E, et al. Phase I safety, pharmacokinetics, and inhibition of SRC activity study of saracatinib in patients with solid tumors. Clin Cancer Res. 2010;16:4876–83.
Park BJ, Whichard ZL, Corey SJ. Dasatinib synergizes with both cytotoxic and signal transduction inhibitors in heterogeneous breast cancer cell lines—lessons for design of combination targeted therapy. Cancer Lett. 2012;320:104–10.
Neves H, Kwok HF. Recent advances in the field of anti-cancer immunotherapy. BBA Clin. 2015;3:280–8.
Miles D, Roché H, Martin M. Phase III multicenter clinical trial of the Sialyl-TN (STn)-keyhole limpet hemocyanin (KLH) vaccine for metastatic breast cancer. Oncologist. 2011;16:1092–100.
Berman D, Korman A, Peck R, et al. The development of immunomodulatory monoclonal antibodies as a new therapeutic modality for cancer: the Bristol-Myers Squibb experience. Pharmacol Ther. 2015;148:132–53.
Khoja L, Butler MO, Kang SP, et al. Pembrolizumab. J Immunother Cancer. 2015;3:36.
Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–57.
Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62.
Montero AJ, Avancha K, Glück S, et al. A cost-benefit analysis of bevacizumab in combination with paclitaxel in the first-line treatment of patients with metastatic breast cancer. Breast Cancer Res Treat. 2012;132:747–51.
Mackey JR, Ramos-Vazquez M, Lipatov O, et al. Primary results of ROSE/TRIO-12, a randomized placebo-controlled phase III Trial evaluating the addition of ramucirumab to first-line docetaxel chemotherapy in metastatic breast cancer. J ClinOncol. 2014;32:1–8.
Luu T, Frankel P, Chung C, et al. Phase I/II trial of vinorelbine and sorafenib in metastatic breast cancer. Clin Breast Cancer. 2014;14:94–100.
Burstein HJ, Elias AD, Rugo HS, et al. Phase II study of sunitinib malate, an oral multitargeted tyrosine kinase inhibitor, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol. 2008;26:1810–6.
Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061–8.
Stalker L, Pemberton J, Moorehead RA. Inhibition of proliferation and migration of luminal and claudin-low breast cancer cells by PDGFR inhibitors. Cancer Cell Int. 2014;14:89.
Hu-Lowe DD, Zou HY, Grazzini ML, et al. Nonclinical antiangiogenesis and antitumor activities of axitinib (AG-013736), an oral, potent, and selective inhibitor of vascular endothelial growth factor receptor tyrosine kinases 1, 2, 3. Clin Cancer Res. 2008;14:7272–83.
Sharma T, Dhingra R, Singh S, et al. Aflibercept: a novel VEGF targeted agent to explore the future perspectives of anti-angiogenic therapy for the treatment of multiple tumors. Mini Rev Med Chem. 2013;13(4):530–40.
Kort A, Durmus S, Sparidans RW, et al. Brain and testis accumulation of regorafenib is restricted by breast cancer resistance protein (BCRP/ABCG2) and P-glycoprotein (P-GP/ABCB1). Pharm Res. 2015;32:2205–16.
Lu J. Palbociclib: a first-in-class CDK4/CDK6 inhibitor for the treatment of hormone-receptor positive advanced breast cancer. J Hematol Oncol. 2015;8:98. doi:10.1186/s13045-015-0194-5.
Peddi PF, Hurvitz SA. Ado-trastuzumab emtansine (T-DM1) in human epidermal growth factor receptor 2 (HER2)-positive metastatic breast cancer: latest evidence and clinical potential. Ther Adv Med Oncol. 2014;6(5):202–9.
Amiri-Kordestani L, Wedam S, Zhang L, et al. First FDA approval of neoadjuvant therapy for breast cancer: pertuzumab for the treatment of patients with HER2-positive breast cancer. Clin Cancer Res. 2014;20:5359–64.
Patel TA, Dave B, Rodriguez AA, et al. Dual HER2 blockade: preclinical and clinical data. Breast Cancer Res. 2014;16:419. doi:10.1186/s13058-014-0419-5.
Geuna E, Montemurro F, Aglietta M, et al. Potential of afatinib in the treatment of patients with HER2-positive breast cancer. Breast Cancer Targets Ther. 2012;4:131–7.
Nabholtz JM, Abrial C, Mouret-Reynier MA, et al. Multicentricneoadjuvant phase II study of panitumumab combined with an anthracycline/taxane-based chemotherapy in operable triple-negative breast cancer: identification of biologically defined signatures predicting treatment impact. Ann Oncol. 2014;25:1570–7.
Baselga J, Gomez P, Greil R, et al. Randomized phase II study of the anti-epidermal growth factor receptor monoclonal antibody cetuximab with cisplatin versus cisplatin alone in patients with metastatic triple-negative breast cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2013;31:2586–92.
Ryan PD, Neven P, Blackwell KL, et al. P1-17-01: figitumumab plus exemestane versus exemestane as first-line treatment of postmenopausal hormone receptor- positive advanced breast cancer: a randomized, open-label phase II trial. Cancer Res. doi:10.1158/0008-5472.SABCS11-P1-17-01.
Gao J, Chesebrough JW, Cartlidge SA, et al. Dual IGF-I/II-neutralizing antibody MEDI-573 potently inhibits IGF signaling and tumor growth. Cancer Res. 2011;71:1029–40.
Sachdev D, Zhang X, Matise I, et al. The type I insulin-like growth factor receptor regulates cancer metastasis independently of primary tumor growth by promoting invasion and survival. Oncogene. 2010;29:251–62.
Robertson JF, Ferrero JM, Bourgeois H, et al. Ganitumab with either exemestane or fulvestrant for postmenopausal women with advanced, hormone-receptor-positive breast cancer: a randomised, controlled, double-blind, phase 2 trial. Lancet Oncol. 2013;14:228–35.
Fagan DH, Uselman RR, Sachdev D, et al. Acquired resistance to tamoxifen is associated with loss of the type I insulin-like growth factor receptor (IGF1R): implications for breast cancer treatment. Cancer Res. 2012;72:3372–80. doi:10.1158/0008-5472.CAN-12-0684.
Wolff AC, Lazar AA, Bondarenko I, et al. Randomized phase III placebo-controlled trial of letrozole plus oral temsirolimus as first-line endocrine therapy in postmenopausal women with locally advanced or metastatic breast cancer. J Clin Oncol. 2013;31:195–202.
Donald P, McDonnell Wardell SE, et al. Oral selective estrogen receptor downregulators (SERDs), a breakthrough endocrine therapy for breast cancer. J Med Chem. 2015;58:4883–7.
Di Leo A, Jerusalem G, Petruzelka L, et al. Results of the CONFIRM Phase III trial comparing fulvestrant 250 mg with fulvestrant 500 mg in postmenopausal women with estrogen receptor positive advanced breast cancer. J Clin Oncol. 2010;28:4594–600.
Lai A, Kahraman M, Govek S, et al. Identification of GDC-0810 (ARN-810), an orally bioavailable selective estrogen receptor degrader (SERD) that demonstrates robust activity in tamoxifen-resistant breast cancer xenografts. J Med Chem. 2015;58:4888–904.
Degorce SL, Bailey A, Callis R, et al. Investigation of (E)–3-[4-(2-Oxo-3-aryl-chromen-4-yl)oxyphenyl]acrylic acids as oral selective estrogen Receptor down-regulators. J Med Chem. 2015;58:3522–33.
Safdari Y, Khalili M, Ebrahimzadehb MA, et al. Natural inhibitors of PI3K/AKT signaling in breast cancer: emphasis on newly-discovered molecular mechanisms of action. Pharmacol Res. 2015;93:1–10.
Lim W, Park J, Lee YH, et al. Subglutinol A, an immunosuppressive a-pyrone diterpenoid from Fusarium subglutinans, acts as an estrogen receptor antagonist. BiochemBiophys Res Commun. 2015;461:507–12.
Maruthanila VL, Poornima J, Mirunalini S. Attenuation of carcinogenesis and the mechanism underlying by the influence of indole-3-carbinol and its metabolite 3,3′-diindolylmethane: a therapeutic marvel. Adv Pharmacol Sci. 2014;. doi:10.1155/2014/832161.
Suzuki N, Liu X, Laxmi YR, et al. Anti-breast cancer potential of SS5020, a novel benzopyran antiestrogen. Int J Cancer. 2011;128:974–82.
Laxmi YR, Liu X, Suzuki N, et al. Anti-breast cancer potential of SS1020, a novel antiestrogen lacking estrogenic and genotoxic actions. Int J Cancer. 2010;127:1718–26.
Buzdar A, Vogel C, Schwartzberg L, et al. Randomized double-blind phase 2 trial of 3 doses of TAS-108 in patients with advanced or metastatic postmenopausal breast cancer. Cancer. 2012;118:3244–53.
Nishino T, Yamanouchi H, Ishibashi K, et al. Antiovulatory effect of a single injection of pure antiestrogen ZK 191703 at early stage of rat estrus cycle. J Steroid Biochem Mol Biol. 2009;114:152–60.
Van de Velde P, Nique F, Bouchoux F, et al. RU 58,668, a new pure antiestrogen inducing a regression of human mammary carcinoma implanted in nude mice. J Steroid Biochem Mol Biol. 1994;48:187–96.
Garner F, Shomali M, Paquin D, et al. RAD1901: a novel, orally bioavailable selective estrogen receptor degrader that demonstrates antitumor activity in breast cancer xenograft models. Anticancer Drugs. 2015;26:948–56.
Acknowledgments
Thanks are attributable to DST &DBT, Govt. of India for financial support and Bioinformatics Infrastructure facility, IASST.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
We declare that we have no conflict of interest.
About this article

Cite this article
Maruthanila, V.L., Elancheran, R., Kunnumakkara, A.B. et al. Recent development of targeted approaches for the treatment of breast cancer. Breast Cancer 24, 191–219 (2017). https://doi.org/10.1007/s12282-016-0732-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12282-016-0732-1