
Abstract
We previously reported a series of amphipathic helices of stapled heptapeptides as membrane-lytic antimicrobial peptides. These peptides possess three lysine residues as the sole cationic amino acid residues in their hydrophilic face of the helix. Lysine-to-arginine substitution is often shown to increase antimicrobial activity of many natural AMPs due to the more favorable interactions of guanidinium moiety of arginine with membranes. In an effort to further improve the pharmacological properties of our novel AMP series, we here examined the impact of lysine-to-arginine substitution on their structures and antimicrobial and hemolytic activities. Our results indicate that the lysine-to-arginine substitution does not always guarantee enhancement in the antimicrobial potency of AMPs. Instead, we observed varied potency and selectivity depending on the number of substitutions and the positions substituted. Our results imply that, in the given helical scaffold stabilized by a hydrocarbon staple, antimicrobial potency and selectivity are influenced by a complex effect of various structural and chemical changes accompanied by lysine-to-arginine substitution rather than solely by the type of cationic residue. These data show potential for use in our scaffold-assisted development of short, selective, and metabolically stable AMPs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Amphipathic helical antimicrobial peptides (AMPs) are a well-known class of the membrane-lytic antibiotics, that may provide an alternative battle strategy against the growing public problem of antibiotic resistance (Shai 2002; Jiang et al. 2008; Huang et al. 2010). Despite their great potential as a novel class of antibiotics, their practical application in clinic has been hampered by some serious drawbacks. Their relatively long sequences require high production cost. Their poor selectivity between prokaryotic and eukaryotic cells causes toxicity. Most importantly, their vulnerability to proteolytic enzymes results in poor bioavailability (Marr et al. 2006). Therefore, various strategies have been studied to complement those weaknesses of AMPs.
In our previous studies, we demonstrated that Verdine’s all-hydrocarbon stapling system (Schafmeister et al. 2000; Kim and Verdine 2009; Kim et al. 2011; Verdine and Hilinski 2012) is a highly useful tool for the development of such short, selective, and metabolically stable AMPs. As part of such effort, we reported a series of stapled heptapeptides as potential AMPs (Dinh et al. 2014, 2015; Luong et al. 2016, 2017). By virtue of the powerful helix-stabilizing effect of oct-4-enyl staple, these heptapeptides were designed to adopt amphipathic α-helices that have a hydrophilic face formed by three lysine residues at positions 1, 4, and 7 and a hydrophobic face formed by the oct-4-enyl hydrocarbon staple on the opposite side (Fig. 1). These stapled heptapeptides exhibited significantly enhanced stability against proteolytic degradation due to their conformational rigidity. They also displayed reasonable antimicrobial activity against both Gram-positive and -negative species of bacteria along with very low hemolytic activity against human red blood cells. In our previous study, we showed that the antimicrobial activity of this series is affected by various structural elements including the introduction of oct-4-enyl staple, position of tryptophan residue, net charge, property of N-terminal capping moiety, and chemical properties of residue at position 5, leading to peptide KKK as the most promising analog in this series (Luong et al. 2017) (Fig. 2a). However, its antimicrobial activity is not potent enough for clinical application and therefore has yet to be improved.

Schematic presentation of the ruthenium-based all-hydrocarbon stapling chemistry that cross-links positions 2 and 6 via an oct-4-enyl tether yielding a stapled heptapeptide helix. Previous studies indicated that the i,i + 4 stapling exclusively yields cis olefin (Bhattacharya et al. 2008; Zhang et al. 2008; Phillips et al. 2011)

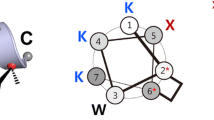
a Helical wheel projection of the stapled heptapeptide KKK. b Sequences of mutated amphipathic heptapeptides studied in this report. The solid line connecting positions 2 and 6 represents the oct-4-enyl hydrocarbon staple. Asterisks in position 2 and 6 represent cross-linked residues. K, NL, R, and W represent lysine, norleucine, arginine, and tryptophan residue, respectively. N-termini of all the peptides are in their free form and their C-termini are amidated
Cationic amino acids are important for the antimicrobial activity of most amphipathic helical AMPs because they mediate the initial interaction with negatively charged bacterial membranes via electrostatic attraction (Yeaman and Yount 2003; Lohner et al. 2008). In the sequence of peptide KKK, three lysine residues were also introduced for this purpose. However, it is well known that arginine is more effective in mediating such peptide-membrane interactions due to the stronger H-bonding capability of its guanidinium moiety compared to the primary amine moiety of lysine (Nguyen et al. 2011; Schmidt and Wong 2013; Li et al. 2013; Arias et al. 2018). Therefore, in this study we investigated the possibility of further improving antimicrobial activity of peptide KKK by substituting each lysine residue at position 1, 4, and 7 with arginine (Fig. 2b).
Materials and methods
General
Fmoc-(S)-α-methyl,α-petenylglycine-OH was purchased from Okeanos Tech Co. Ltd (Beijing, China). All other Fmoc-protected α-amino acids, Rink Amide MBHA resin, and 1-[(1-(cyano-2-ethoxy-2-oxoethylideneaminooxy)-dimethylamino-morpholino)]uranium hexafluorophosphate (COMU) were purchased from NovaBiochem (San Diago, CA, USA). Dimethylformamide (DMF), dichloromethane (DCM), piperidine, Grubbs 1st generation catalyst (bis(tricyclohexylphosphine)benzylidine ruthenium (IV) dichloride), N,N-diisopropylethylamine (DIEA), triisopropylsilane (TIS), N-methyl-2-pyrrolidinone (NMP), trifluoroacetic acid (TFA), and 1,2-dichloroethane (DCE) were purchased from Sigma-Aldrich (St. Louis, MO, USA). All the commercially available reagents and solvents were used as received.
Peptide synthesis
Peptides were prepared using the published protocol (Kim et al. 2011). Briefly, Rink Amide MBHA resin (loading capacity = 0.6 mmol/g) was swelled in NMP for 10 min. The Fmoc group was eliminated using 25% piperidine in NMP (2 × 10 min). Peptide elongation was performed by treating the resin with a mixture of Fmoc-protected amino acid (5 equiv.), COMU (4.75 equiv.), and DIEA (10 equiv.) in NMP for 30 min. For the coupling of Fmoc-(S)-α-methyl,α-petenylglycine, 3 equiv. of Fmoc-protected amino acid was treated with COMU (2.85 equiv.) and DIEA (6 equiv.) for 2 h. The resin was washed with DCM (1 × 2 min), NMP (1 × 2 min), DCM (1 × 2 min), and NMP (1 × 2 min) after each coupling or deprotection reaction.
Metathesis and purification
Ring-closing metathesis (RCM) of protected peptides was performed on the solid support using 20 mol% of Grubbs I catalyst in degassed DCE at room temperature for 2 h. The RCM reaction was repeated using a fresh solution of Grubbs I catalyst for 2 h. After the resin was washed with DCE (3 × 2 min) and DCM (3 × 2 min), the Fmoc group was removed by treating with 25% piperidine in NMP (2 × 10 min). The resin was washed with DCM (3 × 2 min) and DMF (3 × 2 min) and dried in vacuo overnight. The peptides were cleaved from the resin by treating with a mixture of TFA/TIS/water (95/2.5/2.5) for 2 h. The products were precipitated by adding a mixture of n-pentane and diethyl ether (1:1). The mixture was centrifuged, and the solvents was carefully decanted from the container to leave a mixture of the peptide products and the resin. After air-dried in 10 min, the peptides were dissolved in 50% acetonitrile in water. After the resin was filtered off, the peptide products were purified by reverse phase HPLC using a Zorbax C18 column (Agilent, 5 µm, 9.4 × 250 mm). The purified peptides were characterized using LC/MS (Shimadzu LCMS-2020).
- KKK :
-
ESIMS m/z for C47H78N12O7 [M+2H]2+/2 calcd 476.33, found 476.55
- RKK :
-
ESIMS m/z for C47H78N14O7 [M+2H]2+/2 calcd 490.33, found 490.35
- KRK :
-
ESIMS m/z for C47H78N14O7 [M+2H]2+/2 calcd 490.33, found 490.60
- KKR :
-
ESIMS m/z for C47H78N14O7 [M+2H]2+/2 calcd 490.33, found 490.50
- RRK :
-
ESIMS m/z for C47H78N16O7 [M+2H]2+/2 calcd 504.34, found 504.60
- RKR :
-
ESIMS m/z for C47H78N16O7 [M+2H]2+/2 calcd 504.34, found 504.55
- KRR :
-
ESIMS m/z for C47H78N16O7 [M+2H]2+/2 calcd 504.34, found 504.60
- RRR :
-
ESIMS m/z for C47H78N18O7 [M+2H]2+/2 calcd 518.34, found 518.60
- GLU :
-
ESIMS m/z for C46H74N12O9 [M+2H]2+/2 calcd 484.31, found 484.60
Circular dichroism
The sample solutions were prepared by dissolving each peptide in a 25 mM potassium phosphate buffer (pH 6.5). Peptide concentrations were determined based on the absorbance of tryptophan residue at 280 mm (λ280 = 5690 cm−1). Circular dichroism (CD) spectra were collected on a Chirascan HP dual polarization CD spectrometer using standard measurement parameters: 1 nm bandwidth, 0.1 cm path length, 3 accumulations, 1 nm step resolution, and 0.5 s response. The CD spectra were converted into a uniform scale of molar ellipticity after background subtraction. The CD curves were smoothed using standard parameters.
Antimicrobial assay
Antimicrobial activity was determined by measuring the minimal inhibitory concentration (MIC) values using the standard broth microdilution method (Jorgensen and Ferraro 1998) with slight modifications. The assay was conducted against three Gram-positive strains (Bacillus subtilis ATCC 6633, Staphylococcus aureus ATCC 6538p, Staphylococcus epidermis ATCC 12228) and five Gram-negative strains (Escherichia coli ATCC 25922, Shigella dysentariae ATCC 9752, Salmonella typhimurium ATCC 14028, Klebsiella pneumonia ATCC 10031, Pseudomonas aeruginosa ATCC 27853). The bacteria were incubated in 2 ml Luria–Bertani broth at 37 °C overnight. The peptide solutions, prepared with twofold dilutions from 0.2 to 200 µg/mL, were mixed with the bacterial inoculums (106–108 colony-forming units (CFU)/mL) in 96-well round-bottom microtiter plates. After 24 h of incubation at 37 °C, each well was examined to determine MICs, the lowest peptide concentrations completely inhibiting cell growth. All the peptides were tested in duplicate. The bacterial strains used in this work were obtained from the Korean Collection for Type Culture at the Korean Research Institute of Bioscience and Biotechnology (Daejeon, Korea).
Hemolysis assay
190 µL of human red blood cell suspensions (10% v/v in PBS) were treated with 10 µl of serially diluted peptides (ranging 0.8–200 µg/ml in PBS) and incubated at 37 °C for 30 min. After collected by centrifugation, the supernatants were diluted by tenfold with PBS and transferred to a new microtiter plate for spectrophotometric readings at 405 nm. 100% hemolysis was determined by treating the blood suspension with 0.2% Triton X-100. % Hemolysis was calculated based on the following equation:
Results
To investigate effects of lysine-to-arginine substitution on the antimicrobial activity of KKK, we prepared seven mutant analogs using a previously reported protocol (Kim et al. 2011) (Fig. 2b). Mono-substitution at each position of 1, 4, and 7 of KKK yielded analogs RKK, KRK, and KKR, respectively. Double substitution at positions 1 and 4, 1 and 7, and 4 and 7 resulted in analogs KRR, RKR, and RRK, respectively. Substitution of all lysine residues of KKK with arginine gave rise to analog RRR.
The secondary structures of the peptide analogs were determined using far ultra-violet circular dichroism (CD) spectroscopy. All analogs displayed similar CD spectra to that of KKK, having two minima near 208 and 222 nm and a maximum near 190 nm, which are characteristic of α-helices (Fig. 3). Judging by the signal intensity at 222 nm (Chen et al. 1972), the parent KKK appeared to be the most helical among the peptides in this panel; all the arginine-substituted analogs were slightly less helical with RRR being the least helical.

Circular dichroism spectra of stapled heptapeptides in a 25 mM potassium phosphate buffer solution at 20 °C
The antimicrobial activities of the mutant peptides were evaluated in vitro against a set of representative bacterial strains including three Gram-positive and five Gram-negative species, using the broth microdilution method previously reported (Table 1). Analog kkk, which is a KKK derivative bearing glutamate in place of norleucine, was also included as a negative control in this assay (Luong et al. 2017). Compared to KKK, analogs RKK, KRR, and RKR displayed roughly twofold higher inhibitory activity against Gram-positive strains and a similar or twofold higher activity against Gram-negative strains. Analog KKR displayed an activity profile similar to KKK against both Gram-positive and –negative pathogens. In contrast, compared to KKK, analogs RRK, RKR, and RRR exhibited an approximately twofold decrease in activity against Gram-negative microbes, although their potency against Gram-positive microbes remained the same. All mutated peptides showed very low hemolytic activity against human red blood cells. Only RKK, KRR, and RKR displayed a slight increase at the given concentrations.
Discussion
Interactions with bacterial membranes are known to be a critical element for the antimicrobial action of most AMPs. Positively charged amino acid residues play a key role during the initial step of complex formation between AMPs and bacterial membranes due to their favorable electrostatic interactions with bacterial membranes. We previously reported a series of amphipathic helices of stapled heptapeptides containing lysine as the sole cationic amino acid residues, which showed reasonable antimicrobial activity against various bacterial species. In an effort to improve pharmacological properties of this series of novel AMPs, we examined the impact of lysine-to-arginine substitution on their structures and antimicrobial and hemolytic activities. We selected analog KKK as the parent control as it was the most active analog in the series (Luong et al. 2017) and systematically substituted its lysine residues with arginine, which yielded seven mutated analogs (Fig. 2).
Positively charged moieties in the side-chain of lysine and arginine residues have different chemical nature. Therefore, the lysine-to-arginine substitution introduced in the sequence of KKK may influence its secondary structures. In the CD analysis, all mutated analogs displayed a slight decrease in helicity compared to the parent KKK (Fig. 3). In particular, RRR was the least helical analog in this panel. This result indicated that lysine-to-arginine substitution has a negative influence on the helical structure of this series of stapled heptapeptides. Three cationic residues at position 1, 4, and 7 would reside on the same face of the helical scaffold formed by the helix-stabilizing oct-4-enyl staple. For this reason, all the stapled heptapeptides would suffer from a strong electrostatic repulsion in such a highly compact space. Arginine has a shorter and more basic side-chain compared to lysine (pKa of the guanidino group of arginine is 12–13.7; the amino group of lysine, ~ 10.5). Therefore, electrostatic repulsion might be more severe when lysine residues are replaced with arginine. Unfavorable torsional strains induced by this stronger electrostatic repulsion in arginine-substituted analogs may be attributed to their slightly decreased helicity. This is most likely why peptide RRR has the lowest helical content in this series of stapled heptapeptides. Nonetheless, due to the strong helix-stabilizing capability of hydrocarbon staple, all mutated analogs still appear to maintain α-helical conformations as the CD data revealed.
Helical content is well known as one of the critical determinants for the antimicrobial activity of many amphipathic helical AMPs (Huang et al. 2010). In our previous study, we demonstrated that helix-stabilization of the heptapeptides via an oct-4-enyl staple significantly promoted their antimicrobial activity. In contrast, their unstapled counterparts were shown to be unstructured and completely inactive against all the bacterial strains tested in the study (Dinh et al. 2014). In this current work, the helical content of the mutated peptides was not directly correlated with the antimicrobial activity; some arginine-substituted analogs showed an increase in inhibitory potency despite a slight decrease in helicity. Overall, lysine-to-arginine substitution in the sequence of KKK did not guarantee the global improvement in inhibitory activity against all bacterial strains tested in this work. Instead, a slight decrease in activity against Gram-negative microbes was observed for some arginine-substituted analogs (KRK, RRK, and RRR); the sensitivity of Gram-positive pathogens to these analogs remained the same as that of KKK. Also, some mutated peptides displayed a noticeable increase in activity against Gram-positive strains (RKK, RKR, and KRR). Their activity against Gram-negative bacteria did not significantly change compared to KKK. Interestingly, peptides that showed increased activity against Gram-positive strains also exhibited an increase in hemolytic activity compared to KKK.
In many cases, lysine-to-arginine substitution in natural AMPs gave rise to an increase in antimicrobial activity due to the more favorable interaction of guanidinium moiety of arginine with membranes. In this study, we examined the possibility of improving antimicrobial activity of KKK, the most active analog among our stapled heptapeptide series, by substituting lysine residues with arginine. Our results indicated that the lysine-to-arginine substitution does not always guarantee enhancement in antimicrobial potency of AMPs. Instead, we observed varied potency and selectivity depending on the number of the substitutions and which positions were substituted. Our results imply that, in the given helical scaffold stabilized by a hydrocarbon staple, antimicrobial potency and selectivity are influenced by a complex effect of various structural and chemical changes accompanied by lysine-to-arginine substitution rather than solely by the type of cationic residue. These data show potential for use in our scaffold-assisted development of short, selective, and metabolically stable AMPs.
References
Arias M, Piga KB, Hyndman ME, Vogel HJ (2018) Improving the activity of Trp-rich antimicrobial peptides by Arg/Lys substitutions and changing the length of cationic residues. Biomolecules. https://doi.org/10.3390/biom8020019
Bhattacharya S, Zhang H, Debnath AK, Cowburn D (2008) Solution structure of a hydrocarbon stapled peptide inhibitor in complex with monomeric C-terminal domain of HIV-1 capsid. J Biol Chem 283(24):16274–16278
Chen Y-H, Yang J-T, Martinez HM (1972) Determination of the secondary structures of proteins by circular dichroism and optical rotatory dispersion. Biochemistry 11:4120–4131
Dinh TTT, Kim D-H, Lee B-J, Kim Y-W (2014) De novo design and their antimicrobial activity of stapled amphipathic helices of heptapeptides. Bull Korean Chem Soc 35:3632–3636
Dinh TTT, Kim D-H, Nguyen TQ, Lee B-J, Kim Y-W (2015) N-Capping effects of stapled heptapeptides on antimicrobial and hemolytic activities. Bull Korean Chem Soc 36:2511–2515
Huang Y, Huang J, Chen Y (2010) Alpha-helical cationic antimicrobial peptides: relationships of structure and function. Protein Cell 1:143–152
Jiang Z, Vasil AI, Hale JD, Hancock REW, Vasil ML, Hodges RS (2008) Effects of net charge and the number of positively charged residues on the biological activity of amphipathic α-helical cationic antimicrobial peptides. Pept Sci 90:369–383
Jorgensen JH, Ferraro MJ (1998) Antimicrobial susceptibility testing: general principles and contemporary practices. Clin Infect Dis 26:973–980
Kim Y-W, Verdine GL (2009) Stereochemical effects of all-hydrocarbon tethers in i, i + 4 stapled peptides. Bioorg Med Chem Lett 19:2533–2536
Kim Y-W, Grossmann TN, Verdine GL (2011) Synthesis of all-hydrocarbon stapled [alpha]-helical peptides by ring-closing olefin metathesis. Nat Proc 26:761–771
Li L, Vorobyov I, Allen TW (2013) The different interaction of lysine and arginine side chains with lipid membranes. J Phys Chem B 117:11906–11920
Lohner K, Sevcsik E, Pabst G (2008) Liposome-based biomembrane mimetic systems: Implications for lipid-peptide interactions. In: Liu AL (ed) Advances in planar lipid bilayers and liposomes, vol 6, 1st edn. Elsevier, Waltham, pp 103–132
Luong HX, Kim D-H, Lee B-J, Kim Y-W (2016) Antimicrobial and hemolytic activity of stapled heptapeptide dimers. Bull Korean Chem Soc 37:1199–1203
Luong HX, Kim D-H, Mai NT, Lee B-J, Kim Y-W (2017) Mono-substitution effects on antimicrobial activity of stapled heptapeptides. Arch Pharm Res 40:713–719
Marr AK, Gooderham WJ, Hancock REW (2006) Antibacterial peptides for therapeutic use: obstacles and realistic outlook. Curr Opin Pharmacol 6:468–472
Nguyen LT, de Boer L, Zaat SA, Vogel HJ (2011) Investigating the cationic side chains of the antimicrobial peptide tritripticin: hydrogen bonding properties govern its membrane-disruptive activities. Biochim Biophys Acta 1808:2297–2303
Phillips C, Roberts LR, Schade M, Bazin R, Bent A, Davies NL, Moore R, Pannifer AD, Pickford AR, Prior SH, Read CM, Scott A, Brown DG, Xu B, Irving SL (2011) Design and structure of stapled peptides binding to estrogen receptors. J Am Chem Soc 133(25):9696–9699
Schafmeister CE, Po J, Verdine GL (2000) An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides. J Am Chem Soc 122:5891–5892
Schmidt NW, Wong GCL (2013) Antimicrobial peptides and induced membrane curvature: geometry, coordination chemistry, and molecular engineering. Curr Opin Solid State Mater Sci 17:151–163
Shai Y (2002) Mode of action of membrane active antimicrobial peptides. Biopolymers 66:236–248
Verdine GL, Hilinski GJ (2012) Stapled peptides for intracellular drug targets. Methods Enzymol 503:3–33
Yeaman MR, Yount NY (2003) Mechanisms of antimicrobial peptide action and resistance. Pharmacol Res 55:27–55
Zhang H, Zhao Q, Bhattacharya S, Waheed AA, Tong X, Hong A, Heck S, Curreli F, Goger M, Cowburn D, Freed EO, Debnath AK (2008) A cell-penetrating helical peptide as a potential HIV-1 inhibitor. J Mol Biol 378(3):565–580
Acknowledgements
This work was supported by the Basic Research Program through the National Research Foundation of Korea (NRF) Grant funded by the Korea government (MSIT) (2015R1D1A1A01060265).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Luong, H.X., Kim, DH., Lee, BJ. et al. Effects of lysine-to-arginine substitution on antimicrobial activity of cationic stapled heptapeptides. Arch. Pharm. Res. 41, 1092–1097 (2018). https://doi.org/10.1007/s12272-018-1084-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-018-1084-5