
Abstract
The salicylic acid derivative 4-tert-butylphenyl salicylate (4-TBPS) possesses anti-inflammatory activity. We demonstrated this and elucidated the mechanisms involved by using the lipopolysaccharide-stimulated Raw 264.7 mouse macrophage model. The 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, western blot, enzyme-linked immunosorbent assay, and reverse transcriptase-polymerase chain reaction were performed to explore 4-TBPS anti-inflammatory activity. We found that 4-TBPS decreased nitric oxide production without cytotoxic effects on macrophages and reduced expression of inducible nitric oxide synthase (iNOS) and cyclooxygenase (COX)-2 in a dose-dependent manner. Additionally, mRNA expressions of iNOS and COX-2 significantly reduced, with concentrations between 1 and 15 µg/ml. Furthermore, 4-TBPS significantly inhibited the production of pro-inflammatory cytokines including tumor necrosis factor-α (TNF-α), interleukin- (IL)-1β, and IL-6. Moreover, mRNA gene expression of TNF-α, IL-1β, and IL-6 was attenuated in a dose-dependent manner. 4-TBPS potently inhibited translocation of nuclear factor-κB (NF-κB) into the nucleus by degrading IκB kinase (IκBα) following its phosphorylation, thereby causing NF-κB to remain inactive. Collectively, our data indicate that 4-TBPS significantly (p < 0.01) targets the inflammatory response of macrophages via inhibition of iNOS, COX-2, TNF-α, IL-1β, and IL-6 through downregulation of the NF-κB pathway. This indicates that 4-TBPS may have therapeutic potential in inflammatory disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Inflammation is a normal physiological and immune response to tissue injury that involves activation of various immune cells like monocytes and macrophages. During injury, inflammation increases blood supply, improves vascular permeability, and enhances the movement of immune cells to the damaged sites. Abnormal regulation of the inflammatory immune response leads to the development of various pathological states including cancers (Yinon and Karin 2011). Macrophages are key inflammatory cells that participate in the inflammation process by producing cytokines like interleukin (IL)-6, IL-1β, and tumor necrosis factor α (TNF-α). They also produce inflammatory mediators like nitric oxide (NO) and prostaglandins (PGs) (Mosser and Xia 2008). In addition to inducing cell and tissue damage, cytokines also activate macrophages in rheumatoid arthritis, chronic hepatitis as well Vane et al. 1994; Kasama et al. 2005; Wolf et al. 2005 Cheon et al. 2006).
Pro-inflammatory enzymes including the inducible nitric oxide synthase (iNOS) and cyclooxygenase (COX)-2, are involved in the pathogenesis of inflammation and cancer (Klimp et al. 2001; Rajnakova et al. 2001; Son et al. 2001; Nose et al. 2002). Excessive production of NO by iNOS is implicated in the pathogenesis of inflammation as well as cancer (Ohshima 2003). The excess NO acts via reactive nitrogen oxide species-mediated reactions, including deamination of DNA bases and DNA strand breaks (Ohshima 2003). PGE2 is another important inflammatory mediator produced from arachidonic acid metabolites by COX-2 (Esposito and Salvatore 2007; Murakami and Ohigashi 2007). Diverse inflammatory stimuli like lipopolysaccharides (LPS) and pro-inflammatory cytokines activate immune cells to further up-regulate inflammatory mediators. Therefore, these stimuli may be useful tools for investigating the molecular mechanisms of potential agents being developed as new anti-inflammatory drugs (Zeilhofer and Kay 2006; Jachak 2007). Transcription factors belong to the nuclear factor-κB (NF-κB) family and exist in most cells as homodimeric and heterodimeric complexes of p50 and p65 subunits (Farrow and Evers 2002). NF-κB is maintained in an inactive state in the cytoplasm by the NF-κB inhibitor protein (I-κB). NF-κB increases the expression of the genes encoding pro-inflammatory cytokines and enzymes like iNOS and COX-2 (Surh et al. 2001). More importantly, accumulating evidence supports the concept that blocking NF-κB is an important strategy for the control of inflammation and cancer (Karin et al. 2002).
Non-steroidal anti-inflammatory drugs (NSAIDs) are widely used in the treatment of acute and chronic inflammatory diseases. However, their long-term use is associated with the major side effect of gastrointestinal diseases. Therefore, researchers have continued to show an interest in screening new biological compounds from various sources. In our current study, we investigated the anti-inflammatory effects of 4-TBPS (Fig. 1), a salicylic acid derivative. There are previous reports on salicylic acid derivatives that are used for medicinal and cosmetic purposes. They are used as antipyretics, vitamins, antioxidants, NSAIDs, food preservatives, bactericidal agents, antiseptics, etc. (Hutchinson 2003; Madan and Jacob 2014). No pharmacological studies have been previously reported on 4-TBPS. Our novel investigation showed that 4-TBPS exerts in vitro anti-inflammatory effects by inhibiting the expression of iNOS and COX-2 in Raw 264.7 cells. Furthermore, it suppresses the activation of the NF-κB signaling pathway. Therefore, 4-TBPS may be a potential therapeutic candidate for the development of regulatory agents against inflammation.

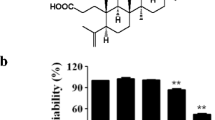
Structure of 4-tert-butylphenyl salicylate
Materials and methods
Materials
The investigational sample of 4-tert-butylphenyl salicylate was obtained from TCI America (Portland, USA). Dulbecco’s Modified Eagle’s Medium (DMEM), fetal bovine serum (FBS), and the penicillin–streptomycin solution were obtained from Invitrogen. LPS (Escherichia coli O11:B4), dimethyl sulfoxide (DMSO), Griess reagent, and 3-(4,5-dimethylthiazol-2-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich (St. Louis, MO, USA). The iNOS, COX-2, NF-κB, IκBα, and p- IκBα polyclonal antibody were obtained from Santa Cruz Biotech Inc. Horseradish peroxidase (HRP)-conjugated donkey anti-rabbit and anti-mouse IgGs were purchased from Cell Signaling. Alkaline phosphatase-conjugated AffiniPure donkey anti-mouse IgG was purchased from Jackson Immunoresearch Laboratories Inc.
Macrophage cell culture
The murine macrophage cell line Raw 264.7, was cultured in DMEM containing 10 % heat-inactivated fetal bovine serum (FBS), 100 unit/ml penicillin, 100 μg/ml streptomycin, and 25 μg/ml amphotericin B, at 37 °C exposed to 5 % CO2. The cells were subcultured every 3 days. Cells were washed twice with fresh medium and stimulated with 1 μg/ml LPS.
Cell viability and NO production assay
Cells were seeded on 96-well plates and treated with 4-TBPS, 24 h later. The viability of cultured cells was determined using the MTT assay. MTT is a pale yellow substrate that is reduced to a dark blue formazan product when incubated with viable, living cells. Briefly, after 24 h incubation with or without 4-TBPS, MTT solution (0.5 mg/ml) was added to each well at 1/10 of the volume of the medium. Cells were incubated at 37 °C for 3 h, and DMSO was added to dissolve the formazan crystals. The absorbance was measured at 590 nm, using an assay reader. Relative cell viability was calculated compared to the viability of the untreated control group.
NO levels in the culture supernatants were measured by the Griess reaction. Raw 264.7 cells (106 cells/ml) were plated on 6-well plates and pretreated with the indicated concentrations of 4-TBPS for 30 min, prior to stimulation with 1 μg/ml of LPS for 24 h. Briefly, the sample supernatants were mixed with equal volumes of Griess reagent (1 % sulfanilamide and 0.1 % naphthylethylenediamine dihydrochloride in 5 % phosphoric acid solution) and then incubated at room temperature for 10 min. The absorbance was measured at 550 nm on a microplate reader (Thermo Co.). Nitrite concentration was determined using a known dilution of sodium nitrite as a standard.
Preparation of nuclear extracts
Nuclear extracts were prepared using a modified method of Wadsworth and Koop (Wadsworth and Dennis 1999). Treated cells were washed, then scraped into 1.5 ml of ice-cold Tris-buffered saline (TBS, pH 7.9), and centrifuged at 12,000 rpm for 30 s. The pellet was suspended in 10 mM N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES, pH 7.9) with 10 mM potassium chloride (KCl), 0.1 mM ethylenediamine tetraacetic acid (EDTA), 0.1 mM ethyleneglycol-bis-β-amini ethyl ether N,N,N′,N′-tetraacetic acid (EGTA), 1 mM dl-dithiothreitol (DTT), 0.5 mM phenylmethylsulfonyl fluoride (PMSF), 5 μg/ml each of leupeptin, aprotinin, and pepstatin. This suspension was incubated on ice for 15 min and then vortexed for 10 s with 0.6 % non-denaturing detergent Nonidet P-40. The nuclei were separated from the cytosol by centrifugation at 12,000 rpm for 60 s. The supernatant was removed, and the pellet was suspended in 50–100 μl of 20 mM HEPES (pH 7.9) containing 25 % glycerol, 0.4 M sodium chloride (NaCl), 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 0.5 mM PMSF, and 10 μg/ml each of leupeptin, aprotinin, and pepstatin. The samples were incubated with rocking at 4 °C for 15 min and then centrifuged for 5 min at 10,000 rpm. The protein concentration of the supernatant was determined using the Bradford assay.
Western blot analysis
Macrophages were incubated with or without LPS, in the presence or absence of 4-TBPS. The cells were harvested, washed twice with ice cold TBS, and resuspended in lysis buffer consisting of 100 mM Tris, 5 mM EDTA, 50 mM NaCl, 50 mM β-glycerophosphate, 50 mM sodium fluoride (NaF), 0.5 % NP-40, 1 % sodium deoxycholate, 0.1 mM sodium orthovanadate, and 1 % PMSF. The cytosolic fraction was obtained from the supernatant after centrifugation at 12,000 rpm for 20 min, at 4 °C. Protein samples (20 μg) were separated using 10 % sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PDVF) membranes. The membranes were blocked with 5 % non-fat milk in TBS–Tween 20 (0.1 %) for 2 h and then incubated with specific primary antibodies for iNOS, COX-2, NF-κB, IκB, and phospho IκB (1:1000, Santa Cruz Biotechnology). Equal loading of the lanes was confirmed by analyzing the blots for β-actin and Lamin B expression using an anti-β-actin and anti-Lamin B antibody (Santa Cruz Biotechnology) respectively. After three washes with TBS–Tween 20, the membrane was hybridized with HRP-conjugated secondary antibody for 1 h. The membranes were washed three times for 10 min and developed with an electrochemiluminescent (ECL) western blotting detection system. The immunoreactive proteins were detected using a LAS-3000 luminescent image analyzer (Fuji Photo Film Co., Ltd.).
Enzyme-linked immunosorbent assay (ELISA) of TNF-α, IL-1β, and IL-6
Inhibition of the production of pro-inflammatory cytokines by 4-TBPS was determined. Cells were treated with different concentrations of 4-TBPS for 1 h, followed by LPS (1 μg/ml) for 24 h. The nitrite content of the supernatant samples was calculated using a standard curve. The supernatant was then analyzed for TNF-α, IL-1β, and IL-6 using enzyme-linked immunosorbent assay (ELISA) with commercial kits (BD Biosciences) according to the manufacturer’s instructions.
RNA preparation and mRNA expression analysis by reverse transcriptase-polymerase chain reaction (RT-PCR)
Total RNA from the 4-TBPS-treated cells was prepared using RNAiso reagent (Takara) according to the manufacturer’s protocol, and stored at −80 °C until use. For the detection of iNOS, COX-2, TNF-α, IL-Iβ, and IL-6 total RNA was extracted, following stimulation and treatment of the cells. The mRNA expression levels of iNOS, COX-2, TNF-α, IL-I β, and IL-6 in the treated cells were compared with control cells.
A total of 1 μg of RNA was reverse-transcribed into cDNA and used as a template for RT-PCR amplification. The primers and amplification conditions are listed in Table 1.
The PCR was performed with a DNA gene cycler and amplification was followed by denaturation at 94 °C for 30 s, then annealing at 58 °C for 30 s, and primer extension at 72 °C for 40 s. PCR products were analyzed on 1 % agarose gels, and bands were visualized using ethidium bromide staining.
NF-κB luciferase activity assay
To measure LPS induced transcriptional activity of NF-κB, NF-κB luciferase activity assay was carried out where Raw 264.7 cells were plated in 24 well plates overnight and transiently transfected with NF-κB luciferase plasmid and pRL-TK plasmid in the presence of Lipofectamine 2000 Reagent (Invitrogen, San Diego, CA) for 4 h. Transfected cells were recovered in FBS for 3 h and exposed to 1 μg/ml LPS in the presence or absence of 4-TBPS for 12 h. Firefly and Renilla luciferase activities in cell lysates were measured using dual luciferase assay system (Promega). The relative luciferase activity was calculated by normalizing firefly luciferase activity to that of Renilla luciferase.
Statistical analysis
Student’s t test and one- way multivariate analysis of variance (ANOVA) were used to analyze the data. Differences were considered significant when p < 0.01.
Results
Cell viability
Raw 264.7 cells were treated with various concentrations of 4-TBPS for 24 h and the cell viability evaluated using an MTT assay. As shown in Fig. 2, 4-TBPS did not exhibit cytotoxicity against Raw 264.7 cells at the range of 1–15 μg/ml. Even at 20 μg/mL, ~60 % of the cells were viable. The 4-TBPS was used at concentrations below 15 μg/ml for further experiments.

Effects of 4-TBPS on cell viability. Cell viability was measured after 24 h incubation. Survival rates were tested with MTT assay in Raw 264.7 cells. Raw 264.7 cells were incubated in the presence or absence of 1-20 µg/ml 4-TBPS for 24 h. Each bar shows the mean ± S.D of three independent experiments performed in triplicate
Effects of 4-TBPS on NO production and iNOS expression in LPS-induced Raw 264.7 macrophage cells
The iNOS enzyme is critical in the pathophysiological process of inflammation and induction of NO. As illustrated in Fig. 3a, b, 4-TBPS inhibited LPS-induced NO production and iNOS expression in Raw 264.7 macrophage cells in a dose-dependent manner. In the absence of LPS, 4-TBPS did not affect NO production and iNOS expression in the cells. Raw 264.7 cells treated with 1 μg/ml of LPS showed a significant increase in levels of NO production and iNOS expression. Western blot, immunofluorescence, and RT-PCR analyzes indicated that treatment with 4-TBPS at 1–20 μg/ml significantly inhibited LPS-induced production of NO, expression of the iNOS protein, and mRNA dose-dependently (Fig. 4a).

Effects of 4-TBPS on LPS-stimulated iNOS and COX-2 expression in Raw 264.7 cells and production of NO. a Inhibition of LPS-stimulated NO production by 4-TBPS in Raw 264.7 cells. The cells were treated with indicated concentrations of 4-TBPS for 1 h prior to the addition of 1 µg/ml of LPS, and the cells were further incubated for 12 h. NO production was determined in culture supernatant by Griess reagent. Results represent the mean ± S.D of three independent experiments performed in triplicate. *p < 0.05; compared to LPS alone. b, Inhibition of LPS-stimulated iNOS expression by 4-TBPS. The cells were treated with indicated concentrations of 4-TBPS for 1 h prior to the addition of 1 µg/ml of LPS, and the cells were further incubated for 24 h. The levels of iNOS protein were monitored. This experiment has been repeated three times with similar observations. Similarly the inhibition of LPS-stimulated COX-2 expression by 4-TBPS. The cells were treated with indicated concentrations of 4-TBPS for 1 h prior to the addition of 1 µg/ml of LPS, and the cells were further incubated for 24 h. The levels of COX-2 protein were monitored. This experiment has been repeated three times with similar observations

4-TBPS inhibits the expression of mRNA. a Effects of 4-TBPS on the mRNA levels of iNOS and COX-2 in LPS-stimulated Raw 264.7 cells. Raw 264.7 cells (1 × 106 cells/ml) were pre-incubated for 12 h, and the cells were stimulated with LPS (1 µg/ml) in the presence of 4-TBPS (1, 5, 10, 15 µg/ml) for 12 h. b Effect of 4-TBPS on LPS-stimulated TNF-α, IL-1β, IL-6 mRNA gene expression After LPS treatment 2-12 h, the levels of TNF-α, IL-1β, IL-6 mRNA were determined by RT-PCR. GAPDH was used as internal control for RT-PCR assays. Each experiment has been repeated three times with similar observations
Effects of 4-TBPS on COX-2 expression in LPS-induced Raw 264.7 macrophage cells
Next, we examined the effects of 4-TBPS on COX-2 levels. Similarly, cells were stimulated with LPS followed by the measurement of COX-2 levels and total mRNA using western blotting and RT-PCR, respectively. The inhibitory effects of 4-TBPS on the levels of COX-2 (Fig. 3b) and mRNA (Fig. 4a) were significant and dose-dependent.
Inhibitory effects of 4-TBPS on the productions of pro-inflammatory cytokines in LPS-induced Raw 264.7 macrophage cells
During the inflammatory process, cytokines such as TNF-α, IL-1β, and IL-6 are produced, and their level indicates the progression of inflammation. As shown in Fig. 5, the LPS-induced release of cytokines was blocked by 4-TBP in a dose-dependent manner, and confirmed by the determination of mRNA gene expression (Fig. 4b). At high concentrations (15 μg/ml) 4-TBPS inhibited the production of cytokines but did not influence the viability of the RAW 264.7 cells (Fig. 2).

Effects of 4-TBPS in the suppression of TNF-α, IL-1β, IL-6 production in LPS-stimulated Raw 264.7 cells. Cells were incubated with the indicated concentrations of 4-TBPS for 30 min before treatment with LPS (1 µg/ml) for 24 h. After incubation for 24 h, the supernatant was collected, and the amounts of proinflammatory cytokines were measured by ELISA assay. Results represent the mean ± S.D. of three independent experiments. *p < 0.05; compared to LPS alone
Effects of 4-TBPS on NF-κB translocation and phosphorylation of IκBα in LPS-induced Raw 264.7 macrophage cells
To assess the effects of 4-TBPS on LPS-induced NF-κB activation, we examined the translocation of NF-κB into the nucleus, using western blot analysis. As demonstrated in Fig. 6a, cytoplasmic levels of p65 were decreased in response to LPS treatment. Conversely, the LPS-induced change in the levels of p65 in the nucleus and cytoplasm, was inhibited in macrophages treated with 4-TBPS. These results suggest that 4-TBPS inhibits NF-κB binding activity by preventing the LPS-induced translocation of p65 to the nucleus. IκBα is a member of a family of cellular proteins that inhibits the NF-κB transcription factor by masking it and thereby, keeping it in inactive state in the cytoplasm. Figure 6b shows that while the level of p-IκBα decreased in response to phosphorylation whereas it increased the level of IκBα in the cytoplasm. This suggests that an increase in the amount of 4-TBPS enhanced the ability of IκBα to maintain NF-κB in an inactive form in the cytoplasm.

Effects of 4-TBPS on LPS-stimulated NF-κB expression that inhibits translocation into nucleus (a) and inhibition IκBα phosphorylation and degradation (b). The cells were separately treated with indicated concentrations of 4-TBPS for 1 h prior to the addition of 1 µg/ml of LPS, and the cells were further incubated for 24 h. c Inhibition of NF-κB luciferase activity by 4-TBPS. Cells were transfected with an NF-κB luciferase construct. Data represent the mean ± SD of three replicates; *p < 0.05, **p < 0.01, significant versus vehicle-treated control
Further study correlated with NF-κB activation was inhibited by 4-TBPS, reporter gene analyzes were carried out. NF-κB luciferase activity was increased when treated with LPS (1 µg/ml, 12 h), and pretreatment of the cell with 1–15 μg/ml 4-TBPS significantly inhibited NF-κB luciferase activity (Fig. 6c).
Discussion
Prolonged dysregulation of pro-inflammatory genes like iNOS and COX-2 leads to chronic inflammation, which is responsible for various pathological diseases including cancers (Murakami and Ohigashi 2007). Consequently, modulation of pro-inflammatory genes in macrophages is an important strategy for the development of new therapeutic agents against inflammatory diseases.
In the present study, we assessed the potential anti-inflammatory functions of 4-TBPS in vitro, by evaluating its effects on LPS-induced changes in the levels of iNOS, COX-2, and NO in RAW 264.7 macrophages cells. In addition, we determine the effects of 4-TBPS on the production of cytokines including TNF-α, IL-1β, and IL-6 in the same cells. Our western blot analysis revealed that 4-TBPS suppressed the LPS-induced expressions of iNOS and COX-2 while it simultaneously enhanced the production of NO (Fig. 3a, b). This result was corroborated by determining the mRNA, using RT-PCR (Fig. 4a). It was further confirmed via cell viable studies that 4-TBPS was not toxic to RAW 264.7 macrophages cells (Fig. 2). It has been established that during the progression of inflammation, stimulated macrophages produces pro-inflammatory cytokines like IL-6, IL-1β, and TNF-α. Researchers have reported that anti-inflammatory compounds inhibit iNOS expression and secretion of pro-inflammatory cytokines (An et al. 2002; Min et al. 2005; Kim et al. 2009). In this study, 4-TBPS inhibited iNOS-dependent NO production. In addition, it also inhibited TNF-α, IL-6, and IL-1β (Fig. 5), iNOS protein expression, and mRNA (Fig. 4a, b) thereby, suggesting strong anti-inflammatory potential.
Moreover, 4-TBPS suppressed the activation of NF-κB by LPS (Fig. 6c) and our data is consistent with the p65 binding activity, which was reduced dose-dependently by 4-TBPS (Fig. 6a). Therefore, our results showed clearly that 4-TBPS inhibited the expression of pro-inflammatory cytokines (Figs. 4b, 5). We established NF-κB as a target of 4-TBPS and, therefore, studied the mechanism of its inactivation. We demonstrated as illustrated in Fig. 7, the possible mode of action of 4-TBPS in a model of LPS-stimulated macrophage cells expressing IκB kinase (IKK). IKK is an enzyme complex that contributes to the propagation of the cellular response to inflammation (Hacker and Karin 2006). IKK specifically phosphorylates the inhibitory IκBα protein (Karin 1999), which disassociates IκBα from NF-κB. The NF-κB then migrates into the nucleus, which is the site for transcription and activates the expression of genes like iNOS and COX-2. Here, we show that 4-TBPS prevents IκBα phosphorylation and degradation (Fig. 6b), thereby inhibiting the nuclear translocation of p65 protein.

In the proposed model of the molecular mechanism, 4-TBPS exerts its anti-inflammatory effects in LPS-stimulated RAW 264.7 cells. The anti-inflammatory functions have been shown show through inactivating IκBα phosphorylation and inhibiting translocation of NF-κB from cytoplasm to nucleus. Ultimately NF-κB pathway is blocked to produce pro-inflammatory enzymes, iNOS, COX-2 and pro-inflammatory cytokines
We investigated the anti-inflammatory effects of 4-TBPS, a salicylic acid derivative. Although numerous salicylic acid derivatives are used for medicinal and cosmetic purposes, to the best of our knowledge, no pharmacological studies have been reported on 4-TBPS. Therefore, our approach to the evaluation of the anti-inflammatory effect of 4-TBPS is novel.
In summary, our findings collectively suggest that 4-TBPS has anti-inflammatory effects on macrophages, which is mediated via the reduction of pro-inflammatory cytokine levels. Furthermore, these effects are associated with NF-κB inactivation, suppression of NF-κB-regulated proteins, and IκBα phosphorylation. However, these effects should be further elucidated using different in vivo models and clinical studies towards their development and utilization as therapeutic agents.
References
An SJ, Pae HO, Oh GS, Choi BM, Jeong S, Jang SI, Oh H, Kwon TO, Song CE, Chung HT (2002) Inhibition of TNF-alpha, IL-1beta, and IL-6 productions and NF-kappa B activation in lipopolysaccharide-activated RAW 264.7 macrophages by catalposide, an iridoid glycoside isolated from Catalpa ovata G. Don (Bignoniaceae). Int Immunopharmacol 2:1173–1181
Cheon HJ, Rho YH, Choi SJ, Lee YH, Song GG, Sohn J, Won NH, Ji JD (2006) Prostaglandin E2 augments IL-10 signaling and function. J Immunol 177:1092–1100
Esposito E, Salvatore C (2007) The role of nitric oxide synthases in lung inflammation. Curr Opin Investig Drugs (London, England: 2000) 8:899–909
Farrow B, Evers BM (2002) Inflammation and the development of pancreatic cancer. Surg Oncol 10:153–169
Hacker H, Karin M (2006) Regulation and function of IKK and IKK-related kinases. Sci Stke 357:12–14
Hutchinson E (2003) An aspirin a day. Nat Rev Cancer 3:552
Jachak SM (2007) PGE synthase inhibitors as an alternative to COX-2 inhibitors. Curr Opin Investig Drugs (London, England: 2000) 8:411–415
Karin M (1999) How NF-kappaB is activated: the role of the IkappaB kinase (IKK) complex. Oncogene 18:6867–6874
Karin M, Yixue C, Florian RG, Zhi-Wei L (2002) NF-κB in cancer: from innocent bystander to major culprit. Nat Rev Cancer 2:301–310
Kasama T, Miwa T, Isozaki T, Odai T, Adachi M, Kunkel S (2005) Neutrophil-derived cytokines: potential therapeutic targets in inflammation. Curr Drug Targets-Inflamm Allergy 4:273–279
Kim EH, Shim B, Kang S, Jeong G, Lee J, Yu YB, Chun M (2009) Anti-inflammatory effects of Scutellaria baicalensis extract via suppression of immune modulators and MAP kinase signaling molecules. J Ethnopharmacol 126:320–331
Klimp AH, Harry H, Claudia K, Ate GJ, Elisabeth GE, Toos D (2001) Expression of cyclooxygenase-2 and inducible nitric oxide synthase in human ovarian tumors and tumor-associated macrophages. Cancer Res 61:7305–7309
Madan RK, Jacob L (2014) A review of toxicity from topical salicylic acid preparations. J Am Acad Dermatol 70:788–792
Min KR, Lee H, Kim BH, Chung EY, Cho SM, Kim Y (2005) Inhibitory effect of 6-hydroxy-7-methoxychroman-2-carboxylic acid phenylamide on nitric oxide and interleukin-6 production in macrophages. Life Sci 77:3242–3257
Mosser DM, Xia Z (2008) Interleukin-10: new perspectives on an old cytokine. Immunol Rev 226:205–218
Murakami A, Ohigashi H (2007) Targeting NOX, INOS and COX-2 in inflammatory cells: chemoprevention using food phytochemicals. Int J Cancer 121:2357–2363
Nose F, Ichikawa T, Fujiwara M, Okayasu I (2002) Up-regulation of cyclooxygenase-2 expression in lymphocytic thyroiditis and thyroid tumors significant correlation with inducible nitric oxide synthase. Am J Clin Pathol 117:546–551
Ohshima H (2003) Genetic and epigenetic damage induced by reactive nitrogen species: implications in carcinogenesis. Toxicol Lett 140:99–104
Rajnakova A, Shabbir M, Goh PM, Ngoi SS (2001) Expression of nitric oxide synthase, cyclooxygenase, and p53 in different stages of human gastric cancer. Cancer Lett 172:177–185
Son HJ, Kim YH, Park D, Kim JJ, Rhee PL, Paik SW, Choi KW, Song SY, Rhee JC (2001) Interaction between cyclooxygenase-2 and inducible nitric oxide synthase in gastric cancer. J Clin Gastroenterol 33:383–388
Surh YJ, Na HK, Lee JY, Keum YS (2001) Molecular mechanisms underlying anti-tumor promoting activities of heat-processed Panax ginseng C.A, Meyer. J Korean Med Sci 16:38–41
Vane JR, Mitchell J, Appleton I, Tomlinson A, Bishop-Bailey D, Croxtall J, Willoughby Derek A (1994) Inducible isoforms of cyclooxygenase and nitric-oxide synthase in inflammation. Proc Natl Acad Sci 91:2046–2050
Wadsworth TL, Dennis RK (1999) Effects of the wine polyphenolics quercetin and resveratrol on pro-inflammatory cytokine expression in RAW 264.7 macrophages. Biochem Pharmacol 57:941–949
Wolf AM, Wolf D, Rumpold H, Ludwiczek S, Enrich B, Gastl G, Weiss G, Herbert T (2005) The kinase inhibitor imatinib mesylate inhibits TNF-α production in vitro and prevents TNF-dependent acute hepatic inflammation. Proc Natl Acad Sci USA 102:13622–13627
Yinon BN, Karin M (2011) Inflammation meets cancer, with NF-[kappa] B as the matchmaker. Nat Immunol 12:715–723
Zeilhofer HU, Brune K (2006) Analgesic strategies beyond the inhibition of cyclooxygenases. Trends Pharmacol Sci 27:467–474
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) Grant funded by the Korean government (MEST) (2010-0029178) and “Cooperative Research Program for Agriculture Science & Technology Development (Project No. PJ01128901)” Rural Development Administration, Republic of Korea.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interests.
Additional information
Yun Hee Choi and Baek Hee Na both authors contributed equally to this work.
Rights and permissions
About this article

Cite this article
Choi, Y.H., Na, B.H., Choi, Y.S. et al. Anti-inflammatory function of 4-tert-butylphenyl salicylate through down-regulation of the NF-kappa B pathway. Arch. Pharm. Res. 39, 429–436 (2016). https://doi.org/10.1007/s12272-015-0679-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-015-0679-3