
Abstract
Two new diphenyl derivatives, named iizukines A (1) and B (2), along with nine known compounds were isolated from coastal saline soil derived fungus Aspergillus iizukae. The structures were determined by extensive spectroscopic analysis. Their cytotoxicities were preliminarily evaluated on HL-60, BEL-7402 and A-549 cell lines by the MTT assay.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Fungi derived from special ecological niches such as craters, hypersaline waters, tropical forests, deserts and deep sea, are prominent as a source of novel bioactive compounds (Blunt et al. 2005, 2011; Peng et al. 2011; Lin et al. 2012). Coastal saline soil, as a unique ecological environment, provides a large unexplored habitat for special fungal strains. Fungi from this habitat may develop unique metabolic mechanisms during the long evolutionary processes under high-salt and high-pH living conditions, and may gain the abilities to produce novel secondary metabolites. Accordingly, we have recently initiated a program to discover bioactive natural products from fungi isolated from this special ecological environment.
Asterric acid and its derivatives refer to the compounds possessing a diphenyl ether structure. They exhibit a wide range of biological activities, such as antifungal, antimicrobial, and cytototoxic activity (Inamori et al. 1983; Hargreaves et al. 2002; Liu et al. 2006; Liao et al. 2012). Asterric acid was discovered as the first nonapeptide endothelin (ET) binding inhibitor from natural sources (Ohashi et al. 1997) and subsequently applied to the studies on biology and medicine (Lee et al. 2002).
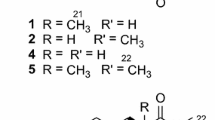
During the course of our investigation on bioactive secondary metabolites of coastal saline soil derived fungi (Ma et al. 2011, 2012), Aspergillus iizukae, which was isolated from coastal saline soil in Kenli, China, attracted our attention. Preliminary studies showed that the culture extract of the strain had strong cytotoxic activity against brine shrimp larvae with IC50 value of 2.7 μg/mL. To the best of our knowledge, there was no report on the secondary metabolites from A. iizukae. Studies on the bioactive extract led to the isolation of two new compounds, named iizukine A (1) and iizukine B (2), together with nine known ones, which were asterric acid (3) (Wu et al. 2008; Liao et al. 2012), methyl chloroasterrate (4) (Hargreaves et al. 2002), methyl dichloroasterrate (5) (Hargreaves et al. 2002), 2, 4-dichloroasterric acid (6) (Curtis et al. 1964; Liao et al. 2012), geodin hydrate (7) (Liao et al. 2012), sulochrin (8) (Ohashi et al. 1997; Lee et al. 2002), monochlorosulochrin (9) (Inamori et al. 1983), questin (10) (Zaman and Khan 2011), and emodin (11) (Ngoc 2008; Zaman and Khan 2011). In this paper, we report the isolation, structure elucidation and cytotoxicity evaluation of all the above compounds.
Materials and methods
General
Melting points were measured on an XRC-1 micro-melting point apparatus and were uncorrected. UV spectra were recorded on a TU-1091 spectrophotometer. IR spectra were taken on a Nicolet 6700 spectrophotometer using attenuated total reflection (ATR) method. NMR spectra were recorded on a Bruker AV-400 spectrometer using TMS as internal standard. HR–ESI–MS was measured on a Q-TOF Ultima GLOBAL GAA076 LC mass spectrometer. Semipreparative HPLC was performed on a Shimadzu LC-6AD Liquid Chromatograph with SPD-20A Detector, using an ODS column [HyperClone 5 μm ODS (C18) 120A, 250 × 10 mm, Phenomenex, 4 mL/min]. All cell lines were purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China).
Fungal material
The working strain, A. iizukae (GenBank accession numbers: HQ717800), was isolated from coastal saline soil in Kenli, Shandong Province of China, in August 2008. The fungus was identified on the basis of sequence analysis of the ITS region of the rDNA. It was deposited in our laboratory.
Fermentation and extraction
Spores were directly inoculated into 500-mL Erlenmeyer flasks containing 200 mL fermentation media (glucose 20 g, maltose 10 g, mannitol 10 g, yeast extract 3 g, KH2PO4 0.5 g, MgSO4·7H2O 0.3 g, dissolved in 1 L sea water). The flasks cultures were incubated at 28 °C on a rotary shaker at 165 rpm for 10 days. Forty liters of whole broth were filtered through cheesecloth to separate the broth supernatant and mycelia. The former was extracted with ethyl acetate, while the latter was extracted with methanol. The methanol extract was evaporated under reduced pressure to afford an aqueous solution, and then extracted with ethyl acetate. The two ethyl acetate extracts were combined and concentrated under reduced pressure to give a crude extract (54 g).
Purification
The crude extract was subjected to a silica gel (200–300 mesh) column packed in petroleum ether eluting with petroleum ether-ethyl acetate and ethyl acetate–methanol in increasing order of polarity to afford eight fractions (Fr1–Fr8). Fr2 was further chromatographed gradiently on silica gel using petroleum ether-ethyl acetate (from 3:1 to 1:5, v/v) to give five subfractions (Fr2-1–Fr2-5). Fr2-3 and Fr2-4 were recrystallized from chloroform to afford compound 10 (12.3 mg) and compound 11 (20.6 mg), respectively. Fr3 was loaded on column chromatography (CC) over silica gel using chloroform–methanol (100:1–10:1, v/v) as mobile phase to give six subfractions (Fr3-1–Fr3-6). Fr3-3 was further purified by Sephadex LH-20 using methanol as eluting solvent to afford compound 3 (8.7 mg). Fr3-4 was rechromatographed over silica gel column using chloroform–methanol (20:1, v/v) as mobile phase, and followed by RP-HPLC with methanol–water-trifluoroacetic acid (60:40:0.005, v/v/v), to yield compound 4 (12.2 mg, t R = 16.2 min) and compound 5 (10.8 mg, t R = 22.1 min). Fr4 was subjected to CC over silica to provide five subfractions (Fr4-1–Fr4-5). Fr4-2 was purified by RP-HPLC using methanol–water-trifluoroacetic acid (65:35:0.005, v/v/v) as eluting solvent to yield compound 2 (10.3 mg, t R = 9.6 min). Fr4-3 was applied to Sephadex LH-20 using methanol as eluting solvent to yield five subfractions (Fr4-3-1-Fr4-3-5). Fr4-3-2, Fr4-3-3, and Fr4-3-4 gave compounds 6 (13.0 mg, t R = 11.2 min), 7 (12.9 mg, t R = 13.6 min) and 1 (9.7 mg, t R = 14.8 min), respectively, after purification by RP-HPLC using methanol–water-trifluoroacetic acid (65:35:0.005, v/v/v) as mobile phase. Fr5 was further separated on Sephadex LH-20 CC with methanol to furnish three subfractions (Fr5-1–Fr5-3). Compound 8 (7.3 mg, t R = 8.1 min) was obtained from Fr5-1 after purification by RP-HPLC using methanol–water-trifluoroacetic acid (70:30:0.005, v/v/v) as mobile phase. Purification of Fr6 by Sephadex LH-20 CC using methanol as the mobile phase gave three subfractions (Fr6-1–Fr6-3). Fr6-3 was further purified using CC over silica gel with chloroform–methanol (20:1, v/v) to give compound 9 (5.3 mg).
Iizukine A (1)
Colorless needles (acetone); mp 143–145 °C; UV (methanol) λ max (log ε): 296 (3.46) nm; IR (ATR) ν max 2,500–3,300, 1,694, 1,606, 1,558, 1,436, 1,403, 1,311, 1,234, 1,183, 1,059, 952, 913, 847, 723 cm−1; HR–ESI–MS m/z 381.0,373 [M−H]− (calcd for C17H14O8Cl, 381.0,372); 1H and 13C NMR data: see Table 1.
Iizukine B (2)
Colorless crystal (acetone); mp 194–196 °C; UV (methanol) λ max (log ε): 284 (3.32) nm; IR (ATR) ν max 3,467, 3,299, 1,599, 1,509, 1,452, 1,423, 1,325, 1,267, 1,231, 1,207, 1,189, 1,083, 1,057, 983, 829, 814, 757 cm−1; HR–ESI–MS m/z 287.1,279 [M−H]− (calcd for C17H19O4, 287.1,278); 1H and 13C NMR data: see Table 1.
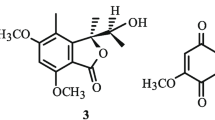
Asterric acid (3)
Colorless needles (acetone); ESI–MS m/z 347 [M−H]−; 1H NMR (400 MHz, DMSO-d 6) δ 13.00 (1H, brs, OH-8′), 11.18 (1H, brs, OH-3′), 9.90 (1H, s, OH-4), 6.78 (1H, s, H-3), 6.78 (1H, s, H-5), 6.33 (1H, s, H-6′), 5.65 (1H, s, H-4′), 3.70 (3H, s, H-7), 3.61 (3H, s, H-9), 2.07 (3H, s, H-7′); 13C NMR (100 MHz, DMSO-d 6) δ 170.3 (C-8′), 165.1 (C-8), 159.8 (C-5′), 158.5 (C-1′), 155.1 (C-4), 153.4 (C-6), 143.2 (C-5′), 133.9 (C-1), 125.3 (C-2), 109.9 (C-4′), 107.5 (C-3), 104.9 (C-6′), 104.7 (C-5), 104.1 (C-2′), 56.1 (C-7), 52.1 (C-9), 21.5 (C-7′).
Methyl chloroasterrate (4)
Colorless needles (acetone); ESI–MS m/z 381, 383 [M−H]−; 1H NMR (400 MHz, DMSO-d 6) δ 13.72 (1H, brs, OH-8′), 12.22 (1H, brs, OH-3′), 9.94 (1H, s, OH-4), 6.80 (1H, brs, H-3), 6.79 (1H, brs, H-5), 5.89 (1H, s, H-6′), 3.70 (3H, s, H-7), 3.62 (3H, s, H-9), 2.17 (3H, s, H-7′); 13C NMR (100 MHz, DMSO-d 6) δ 170.9 (C-8′), 164.9 (C-8), 157.2 (C-1′), 156.4 (C-3′), 155.3 (C-4), 153.2 (C-6), 141.6 (C-5′), 133.7 (C-1), 125.1 (C-2), 113.3 (C-6′), 107.6 (C-3), 106.2 (C-4′), 105.0 (C-5), 104.3 (C-2′), 56.1 (C-7), 52.1 (C-9), 20.6 (C-7′). Crystallographic data of 4 have been deposited in the Cambridge Crystallographic Data Center (no. CCDC 963750). These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Methyl dichloroasterrate (5)
Colorless crystal (acetone); ESI–MS m/z 429, 431, 433 [M−H]−; 1H NMR (400 MHz, DMSO-d 6) δ 9.89 (1H, s, OH-3′), 9.83 (1H, s, OH-4), 6.76 (1H, d, J = 2.6 Hz, H-3), 6.66 (1H, d, J = 2.6 Hz, H-5), 3.65 (3H, s, H-9), 3.61 (3H, s, H-7), 3.27 (3H, s, H-9), 2.45 (3H, s, H-7′); 13C NMR (100 MHz, DMSO-d 6) δ 164.5 (C-8), 163.8 (C-8′), 154.7 (C-4), 152.9 (C-6), 149.2 (C-1′), 149.0 (C-3′), 135.7 (C-5′), 135.1 (C-1), 124.4 (C-2), 115.6 (C-4′), 115.5 (C-6′), 112.4 (C-3′), 107.4 (C-3), 104.7 (C-5), 56.1 (C-7), 52.0 (C-9), 51.9 (C-9′), 18.1 (C-7′). Crystallographic data of 5 have been deposited in the Cambridge Crystallographic Data Center (no. CCDC 963591). These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
2, 4-dichloroasterric acid (6)
Colorless crystal (acetone); ESI–MS m/z 415, 417, 419 [M−H]−; 1H NMR (400 MHz, DMSO-d 6) δ 18.41 (1H, s, OH-8′), 9.28 (1H, s, OH-4), 6.51 (1H, s, H-3), 6.51 (1H, s, H-5), 3.54 (3H, s, H-9), 3.45 (3H, s, H-7), 2.35 (3H, s, H-7′); 13C NMR (100 MHz, DMSO-d 6) δ 166.8 (C-8′), 166.1 (C-8), 160.5 (C-1′), 152.7 (C-3′), 151.4 (C-4), 150.7 (C-6), 139.4 (C-1), 134.9 (C-5′), 122.2 (C-2), 114.6 (C-4′), 112.8 (C-6′), 110.8 (C-3′), 107.2 (C-3), 105.1 (C-5), 56.5 (C-7), 51.4 (C-9), 18.0 (C-7′).
Geodin hydrate (7)
Colorless needles (acetone); ESI–MS m/z 415, 417, 419 [M−H]−; 1H NMR (400 MHz, DMSO-d 6) δ 12.67 (1H, s, OH-8), 9.79 (1H, s, OH-3′), 9.79 (1H, s, OH-4), 6.77 (1H, d, J = 2.7 Hz, H-3), 6.62 (1H, d, J = 2.7 Hz, H-5), 3.60 (H-7), 3.27 (H-9′), 2.44 (H-7′); 13C NMR (100 MHz, DMSO-d 6) δ 165.5 (C-8), 163.8 (C-8′), 154.6 (C-4), 152.8 (C-6), 149.3 (C-1′), 149.0 (C-3′), 135.5 (C-5′), 135.0 (C-1), 125.7 (C-2), 115.5 (C-4′), 115.2 (C-6′), 112.4 (C-2′), 107.7 (C-3), 104.2 (C-5), 56.1 (C-7), 52.0 (C-9′), 18.2 (C-7′).
Sulochrin (8)
Pale yellow needles (acetone); ESI–MS m/z 331 [M−H]−; 1H NMR (400 MHz, DMSO-d 6) δ 11.42 (1H, s, OH-6′), 11.42 (1H, s, OH-2′), 9.96 (1H, s, OH-4), 6.90 (1H, d, J = 1.3 Hz, H-3), 6.67 (1H, d, J = 1.3 Hz, H-5), 6.08 (1H, s, H-3′), 6.08 (1H, s, H-5′), 3.64 (H-7), 3.63 (H-9), 2.15 (H-7′); 13C NMR (100 MHz, DMSO-d 6) δ 199.6 (C-10), 165.6 (C-8), 161.6 (C-2′), 161.6 (C-6′), 158.1 (C-4), 156.8 (C-6), 147.3 (C-4′), 127.9 (C-2), 126.2 (C-1), 109.1 (C-1′), 107.5 (C-3′), 107.5 (C-5′), 107.2 (C-3), 103.4 (C-5), 55.9 (C-7), 52.0 (C-9), 21.5 (C-7′).
Monochlorosulochrin (9)
Pale yellow needles (acetone); ESI–MS m/z 365, 367 [M−H]−; 1H NMR (400 MHz, DMSO-d 6) δ 13.39 (1H, s, OH-6′), 10.45 (1H, s, OH-2′), 10.07 (1H, s, OH-4), 6.92 (1H, d, J = 1.4 Hz, H-3), 6.70 (1H, d, J = 1.4 Hz, H-5), 6.20 (1H, s, H-5′), 3.65 (H-7), 3.65 (H -9), 2.25 (H-7′); 13C NMR (100 MHz, DMSO-d 6) δ 200.1 (C-10), 165.6 (C-8), 158.47 (C-2′), 158.45 (C-6′), 158.3 (C-4), 156.8 (C-6), 144.6 (C-4′), 127.9 (C-2), 125.5 (C-1), 110.4 (C-3′), 110.0 (C-1′), 108.5 (C-5′), 107.2 (C-3), 103.5 (C-5), 56.0 (C-7), 52.2 (C-9), 20.6 (C-7′).
Questin (10)
Pale orange needles (acetone); ESI–MS m/z 283 [M−H]−; 1H NMR (400 MHz, DMSO-d 6) δ 13.22 (1H, s, OH-1), 11.24 (1H, s, OH-6), 7.41 (1H, s, H-4), 7.20 (1H, d, J = 2.2 Hz, H-5), 7.12 (1H, s, H-2), 6.84 (1H, d, J = 2.2 Hz, H-7), 3.90 (3H, s, OCH3), 2.38 (3H, s, CH3); 13C NMR (100 MHz, DMSO-d 6) δ 186.3 (C-9), 182.2 (C-10), 164.4 (C-8), 163.4 (C-1), 161.6 (C-6), 146.6 (C-3), 136.7 (C-10a), 132.0 (C-4a), 124.1 (C-4), 119.1 (C-2), 114.3 (C-9a), 112.6 (C-8a), 106.9 (C-5), 104.9 (C-7), 56.3 (OCH3), 21.3 (CH3).
Emodin (11)
Red needles (methanol); ESI–MS m/z 269 [M−H]−; 1H NMR (400 MHz, DMSO-d 6) δ 12.04 (1H, s, OH-8), 11.96 (1H, s, OH-1), 11.38 (1H, brs, OH-6), 7.42 (1H, H-4), 7.11 (1H, H-2), 7.06 (1H, d, J = 2.1 Hz, H-5), 6.56 (1H, d, J = 2.1 Hz, H-7), 2.38 (3H, s, CH3); 13C NMR (100 MHz, DMSO-d 6) δ 189.6 (C-9), 181.2 (C-10), 165.5 (C-3), 164.4 (C-1), 161.3 (C-8), 148.2 (C-6), 135.0 (C-4a), 132.7 (C-10a), 124.0 (C-7), 120.4 (C-5), 113.2 (C-8a), 108.8 (C-4), 108.7 (C-9a), 107.8 (C-2), 21.5 (CH3).
Cytotoxicity assay in vitro
Compounds 1–11 were subjected to cytotoxic evaluation against HL-60, BEL-7402 and A-549 cell lines with MTT method. Cell lines were cultured in RPMI-1640 medium supplemented with 10 % FBS under a humidified atmosphere of 5 % CO2 and 95 % air at 37 °C. Cell suspensions at a density of 5 × 104 cell/mL were planted in 96 well microtiter plates (200 μL per well) and incubated for 24 h. Then, different concentrations of compounds were added to each well and incubated for another 72 h. After treatment, 20 μL MTT solution (5 mg/mL) was added to each well and incubated for 4 h. The crystals were then dissolved in 100 μL DMSO. Absorbance was recorded on a SPECTRA MAX PLUS plate reader (Molecular Devices, Sunnyvale, CA, USA) at 490 nm.
Results and discussion
All compounds (Fig. 1) were isolated by various chromatographic techniques. Their structures were elucidated from the spectroscopic data, including UV, IR, 1D and 2D NMR, and MS. The structures of 4 and 5 were further proved by single-crystal X-ray diffraction analysis (Fig. 2).

Structures of 1–11

X-ray structures of 4 and 5
Compound 1 was obtained as colorless needles. Its molecular formula was established as C17H15ClO8 with 10 degrees of unsaturation from HR−ESI−MS and 1D-NMR spectroscopic data. The ratio of [M−H]− isotopic peaks (382:384/3:1) clearly indicated the presence of one chlorine atom. The UV spectrum of compound 1 displayed maximum absorption bands at λ max 296 nm. The IR spectrum showed absorption bands in consistent with carboxylic acid (2,500–3,300 cm−1), and carbonyl group (1,694 cm−1). The 1H and 13C NMR spectra disclosed the presence of three methyls (one aromatic, the others oxygenated), three aromatic methines, nine aromatic quaternary carbons (δ C 154.2, 153.2, 152.8, 150.7, 137.7, 135.3, 125.7, 113.4 and 110.4), and two carbonyl signals (δ C 165.5, 164.5). The above information revealed the presence of two phenyl rings (one pentasubstituted, another tetrasubstituted) in 1. Carful comparison of the NMR data for compound 1 and 4 revealed they were almost identical, except the two carbonyls (δ C 165.5 and 164.5 in 1; 164.9 and 170.9 in 4), which suggested they were isomers. The HMBC correlations from H-3 to C-8, and from H-9′ to C-8′ (164.5) suggested the carboxyl group was anchored at C-2, while the ester carbonyl at C-2′ (Fig. 3). The assignments of NMR data for compound 1 were completed with assistance from the HMBC and HMQC correlations.

Key HMBC correlations in compounds 1 and 2
Compound 2 was obtained as colorless crystals. The high resolution electrospray ionization mass spectra indicated a molecular formula of C17H20O4 with 8 degrees of unsaturation. The UV spectrum of compound 2 displayed maximum absorption band at λ max 284 nm. The IR absorption bands at 1,599, 1,509 and 1,452 cm−1 suggested the presence of typical phenyl rings. Analysis of the 1H NMR data in conjunction with the HSQC spectrum disclosed the presence of four singlet methyls (δ H 1.88, 1.90, 2.04, and 2.12), one singlet methylene (δ H 3.74), and two aromatic singlet methines (δ H 6.17, and 6.32). The 13C NMR further displayed 10 aromatic quaternary carbons (δ C 153.7, 153.4, 152.2, 151.1, 137.8, 134.1, 115.6, 114.5, 112.9, 112.9), four of which should be oxygenated in view of their chemical shifts. The above evidence revealed the presence of two benzene rings. The methylene protons resonating downfield at δ H 3.74 (2H, s) suggested that it should link with two phenyl rings, which was confirmed by the HMBC correlations from H-9 to C-1 (δ C 112.9), C-2 (δ C 153.7), C-6 (δ C 152.1), C-1′ (δ C 115.6), C-2′ (δ C 137.8) and C-6′ (δ C 151.1) (Fig. 3). The HMBC correlations from OH-4 to C-1, C-2 and C-3, from H-7 to C-2, C-3 and C-4, from OH-6 to C-5 and C-6, and from H-5 to C-1, C-3, C-4 and C-6 constructed one pentasubstituted phenyl ring. Another pentasubstituted phenyl ring was established from the HMBC correlations from H-7′ to C-1′, C-2′ and C-3′, from H-8′ to C-2′, C-3′ and C-4′, from OH-4′ to C-3′, C-4′ and C-5′, from H-6′ to C-1′, C-5′ and C-6′, and from H-5′ to C-1′, C-3′, C-4′ and C-6′. So the structure of 2 was finally completed as shown in Fig. 1.
The cytotoxicities of all compounds against HL-60 (human promyelocytic leukemia), BEL-7402 (human hepatoma) and A-549 (human lung carcinoma) were tested by the MTT (Mosmann, 1983) assay in vitro (Table 2) with 5-fluorouracil (5-FU) as positive control. As shown in Table 2, all compounds showed weak cytotoxic activities.
References
Blunt, J.W., B.R. Copp, M.H. Munro, P.T. Northcote, and M.R. Prinsep. 2005. Marine natural product. Natural Products Reports 22: 15–61.
Blunt, J.W., B.R. Copp, M.H. Munro, P.T. Northcote, and M.R. Prinsep. 2011. Marine natural products. Natural Products Reports 28: 196–268.
Curtis, R.F., P.C. Harries, C.H. Hassall, and J.D. Levi. 1964. The biosynthesis of phenols. Biochemical Journal 90: 43–51.
Hargreaves, J., J. Park, E.L. Ghisalberti, K. Sivasithamparam, B.W. Skelton, and A.H. White. 2002. New chlorinated diphenyl ethers from an Aspergillus species. Journal of Natural Products 65: 7–10.
Inamori, Y., Y. Kato, M. Kubo, T. Kamiki, T. Takemoto, and K. Nomoto. 1983. Studies on metabolites produced by Aspergillus terreus var. aureus. I. Chemical structures and antimicrobial activities of metabolites isolated from culture broth. Chemical and Pharmaceutical Bulletin 31: 4543–4548.
Lee, H.J., J.H. Lee, B.Y. Hwang, H.S. Kim, and J.J. Lee. 2002. Fungal metabolites, asterric acid derivatives inhibit vascular endothelial growth factor (VEGF)-induced tube formation of HUVECs. Journal of Antibiotics 55: 552–556.
Liao, W.Y., C.N. Shen, L.H. Lin, Y.L. Yang, H.Y. Han, J.W. Chen, S.C. Kuo, S.H. Wu, and C.C. Liaw. 2012. Asperjinone, a nor-neolignan, and terrein, a suppressor of ABCG2-expressing breast cancer cells, from thermophilic Aspergillus terreus. Journal of Natural Products 75: 630–635.
Lin, X.P., X.F. Zhou, F.Z. Wang, K.S. Liu, B. Yang, X.W. Yang, J. Peng, J. Liu, Z. Ren, and Y.H. Liu. 2012. A new cytotoxic sesquiterpene quinone produced by Penicillium sp. F00120 isolated from a deep sea sediment sample. Marine Drugs 10: 106–115.
Liu, R., W.M. Zhu, Y.P. Zhang, T.J. Zhu, H.B. Liu, Y.C. Fang, and Q.Q. Gu. 2006. A new diphenyl ether from marine-derived fungus Aspergillus sp. B-F-2. Journal of Antibiotics 59: 362–365.
Ma, L.Y., W.Z. Liu, Y.L. Huang, and X.G. Rong. 2011. Two acid sorbicillin analogues from saline lands-derived fungus Trichoderma sp. Journal of Antibiotics 64: 645–647.
Ma, L.Y., W.Z. Liu, L. Shen, Y.L. Huang, X.G. Rong, Y.Y. Xu, and X.D. Gao. 2012. Spiroketals, isocoumarin, and indoleformic acid derivatives from saline soil derived fungus Penicillium raistrickii. Tetrahedron 68: 2276–2282.
Mosmann, T.J. 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. Journal of Immunological Methods 65: 55–63.
Ngoc, T.M., PTHong Minh, T.M. Hung, P.T. Thuong, I. Lee, B. Min, and K. Bae. 2008. Lipoxygenase inhibitory constituents from rhubarb. Archives of Pharmacal Research 31: 598–605.
Ohashi, H., M. Ishikawa, J. Ito, A. Ueno, G.J. Gleich, H. Kita, H. Kawai, and H. Fukamachi. 1997. Sulochrin inhibits eosinophil degranulation. Journal of Antibiotics 50: 972–974.
Peng, X.P., Y. Wang, P.P. Liu, K. Hong, H. Chen, X. Yin, and W.M. Zhu. 2011. Aromatic compounds from the halotolerant fungal strain of Wallemia sebi PXP-89 in a hypersaline medium. Archives of Pharmacal Research 34: 907–912.
Wu, S.H., Y.W. Chen, S. Qin, and R. Huang. 2008. A new spiroketal from Aspergillus terreus, an endophytic fungus in Opuntia ficusindica Mill. Journal of Basic Microbiology 48: 140–142.
Zaman, K., M.R. Khan, M. Ali, and D.J. Maitland. 2011. New anthraquinone dimer from the root bark of Cassia artemisioides (Gaudich. Ex. DC) Randell. Journal of Asian Natural Products Research 13: 62–67.
Acknowledgments
This work was supported by the National Natural Science Fund of China (No. 31270082), and Shandong Provincial Natural Science Foundation of China (No. Y2008B17).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Liu, D., Yan, L., Ma, L. et al. Diphenyl derivatives from coastal saline soil fungus Aspergillus iizukae . Arch. Pharm. Res. 38, 1038–1043 (2015). https://doi.org/10.1007/s12272-014-0371-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-014-0371-z