
Abstract
The saponin astragaloside IV (AST) is one of major active components purified from Astragalus membranaceus (Fisch) Bge, which has been used in traditional Chinese medicine to treat immune disorders including rheumatoid arthritis (RA). The effects of AST on the suppression of experimental arthritis and its possible mechanisms are unknown. We measured the paw swelling of ankle joints, splenocyte proliferation, interleukin 1β (IL-1β), tumor necrosis factor α (TNFα) and nitric oxide (NO) formation by macrophages in rat adjuvant-induced arthritis (AIA). Intraarticular injection of IL-1β to rat knee joint for inducing the edema and in vitro IL-1β-stimulated cartilage impairment were examined. The results showed that oral treatment of AST (100 mg/kg/day) suppressed the joint inflammation and inhibited IL-1β, TNFα and NO production in macrophages from AIA rats. Macrophages were one of AST targeted cells, and mediated the reduced splenocyte proliferation in AIA rats. In addition, AST reduced the swelling induced by intraarticular injection of IL-1β, and protected against IL-1β-induced damage of cartilage proteoglycan synthesis and chondrocyte proliferation. We conclude that AST possesses antiarthritic effect and prevents IL-1β-induced joint inflammation and cartilage destruction. These findings suggest that AST may be used for the treatment of RA and other inflammatory joint diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune inflammatory disease characterized by synovial inflammation, articular cartilage impairment and extra-articular manifestations (Davignon et al. 2013; Walter et al. 2013). Many different cell components are involved in the RA development, including neutrophils, T and B lymphocytes, and monocytes/macrophages (Walter et al. 2013; Jovanovic et al. 2011). Activation of these cells leads to the production of cytokines and mediators responsible for inflammation. The important roles of inflammatory cytokines and mediators, such as tumor necrosis factor α (TNFα), interleukin 1 (IL-1), and nitric oxide (NO) were well documented in the pathogenesis of RA (Davignon et al. 2013; Prati et al. 2012; Bao et al. 2012). Recent findings show that keeping an adequate balance between regulatory T cells (Treg) and T helper type 17 (Th17) cells is critical for preventing the development of RA and other inflammatory diseases (Ju et al. 2012; Nistala and Wedderburn 2009). IL-17 synergizes with TNFα in the induction of proinflammatory cytokines and destruction of cartilage and bone (Van Bezooijen et al. 2002). IL-1β plays a critical role in the conversion of human Treg cells into IL-17-producing cells in vitro (Walter et al. 2013; Deknuydt et al. 2009). Treatment with TNFα receptor antagonist has led to successful control of RA, but there are some inadequate responses in a proportion of RA patients (Zivojinovic et al. 2012; Chen et al. 2011). In addition, inactivation of TNFα may interfere with innate immune defense and predispose a risk of pathogenic infection. Therefore, therapy on modulating the function of monocytes/macrophages and lymphocytes is a complementary strategy that may be useful for the treatment of RA and other inflammatory joint diseases.
Astragalus membranaceus (Fisch) Bge is one of the most widely used traditional Chinese tonic herbs presented either as a single herb or as a collection of herbs in a complex prescription. It possesses many biological functions including immunomodulating effects, which has long been used in traditional Chinese medicine to treat autoimmune diseases including RA (Yu et al. 2007; Cho and Leung 2007; Lei et al. 2003; Wang et al. 2002). The extracts of Astragalus membranaceus (Fisch) Bge contain various saponins, polysaccharides, and flavonoids (Yu et al. 2007; Ma et al. 2002). Li and colleagues reported that therapeutic effects of astragalus polysaccharides were related to their inhibition of TNFα and IL-1β production and synovial apoptosis in rats with adjuvant-induced arthritis (Jiang et al. 2010; Li et al. 2009; Yang et al. 2009). In addition, astragalus polysaccharides were shown to induce the differentiation of splenic dendritic cells by shifting of Th2 to Th1 with enhancement of T lymphocyte immune function in vitro (Liu et al. 2011). Studies on pharmacology have demonstrated that the saponin Astragaloside IV (AST) is one of major active components purified from Astragalus membranaceus (Fisch) Bge (Ma et al. 2002; Lei et al. 2003; Wang et al. 2002). In the present studies, we evaluated AST effects on the suppression of experimental arthritis and inflammatory mediator-induced cartilage destruction.
Materials and methods
Animals
Male Sprague-Dawley rats (2–3 months old, 160 ± 20 g) and Male New Zealand White rabbits (2.2–2.8 kg) were provided by the Animal Center of Anhui Institute of Medical Sciences. Animals were kept under a 12:12 h light:dark cycle and allowed free access to food and water. Animals were acclimated to this environment for 7 days prior to treatments. All procedures used in these animal studies were approved by local Administration Office Committee of Laboratory Animal.
Induction of rat adjuvant-induced arthritis (AIA)
Arthritis was induced as previous described (Hu et al. 2012). Briefly, the rats were immunized by a single intradermal injection into the left hind foot pad with 1 mg heat-inactive BCG (Shanghai Biochemical Factory, Shanghai, China) suspended in 0.1 ml paraffin oil (Freund’s complete adjuvant). After immunization, arthritic rats were randomly divided into different groups as indicated. The hind paw volume in both injected and non-injected side was measured by a MK-550 volume meter (Muromachi-Kikai Co, Japan) before and after immunization.
Drug preparation
AST was provided by Jiangsu Institute of Materia Medica, Nanjing, China (purity above 98 % by high-performance liquid chromatography analysis). The chemical structure of AST (C41H68O14, molecular weight = 784.9) was described in Fig. 1. AST was dissolved in 1 % sodium carboxymethylcellulose (CMC-Na) for oral administration.

Chemical structure of Astragaloside IV (C41H68O14, molecular weight = 784)
Dosing regimen
After rats were immunized by injection of Freund’s complete adjuvant on day 0, the AIA rats were orally treated with 0.5 ml of vehicle (CMC-Na), AST (100 mg/kg/day), l-NAME (180 mg/kg/day) and/or l-arginine (540 mg/kg/day) once a day from day 0 to day 27 as indicated to determine the effect of AST and other reagents on the primary and secondary inflammation, respectively.
Intraarticular injection of IL-1β-induced swelling of rat knee joints
The hind legs of rat were shaved and prepared using routine aseptic techniques. All intraarticular injections were performed under ether anesthesia. The rats were given 50 μl recombinant human IL-1β (rhIL-1β) by intraarticular injection to the right hind knee joint (1 μg/knee) on days 0, 2 and 4 to elicit an inflammatory response. An equal volume of sterile physiological saline was injected into the left knee as a control. At the end of the experiment (day 5), the animals were sacrificed and the volume of both knee joints in each rat was measured as described previously (Presle et al. 1999).
Isolation of nonadherent splenocytes and peritoneal macrophages
Spleen was removed aseptically from each rats and transferred to six-well culture plates containing RPMI 1640 medium supplemented with 10 % newborn bovine serum and penicillin (100 units/ml)–streptomycin (100 μg/ml) (Wang and Chen 1998; Wang et al. 1998). The spleen was minced and passed through stainless steel mesh to obtain single cell suspensions. The erythrocytes were lysed with distilled water. The remaining cell pellet was resuspended in medium, which represented the total splenic mononuclear cell population. The nonadherent splenocytes were prepared by incubating the whole splenic mononuclear cell preparation (107 cells/ml) in 10-cm culture dishes at 37 °C in an atmosphere of 5 % CO2 for 24 h to facilitate cell adherence. The nonadherent lymphocyte population was harvested, washed, and resuspended in fresh medium.
Rat peritoneal lavage with cold Hanks’ solution (calcium and magnesium free) was collected and resuspened in RPMI 1640 medium at a concentration of 2 × 106 cells/ml. The cell suspension was seeded in 10-cm culture dishes. After incubation at 37 °C in 5 % CO2 for 3 h, nonadherent cells were removed by gently washing twice with RPMI 1640 medium. The resultant cells were peritoneal macrophages. The toxicity of AST on macrophages was measured by colorimetric MTT ((3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay (Wang and Chen 1998).
Splenocyte proliferation
The splenocytes were reconstituted by adding 5 × 106/ml nonadherent lymphocytes and 1.25 × 105 peritoneal macrophages into 96-well plates (Wang and Chen 1998). The cultures were then incubated at 37 °C for 48 h. [3H]-labeled thymidine (3H-TdR) (0.4 μCi/well) was added for additional 6 h before the termination of incubation. The cells were harvested onto glass fiber paper by a multiwall cell harvester. The radioactivity was measured by FJ-2107 liquid scintillation counter (Xian 262 Factory, China). The results were expressed as means of cpm of triplicate wells.
Preparation of cell culture supernatants
The peritoneal macrophages in 1 ml RPMI 1640 medium containing lipopolysaccharide (10 μg/ml) and/or different concentrations of AST as indicated were seeded in 24-well culture plates. The plate was incubated at 37 °C in an atmosphere of 5 % CO2 for 48 h. The cell culture supernatants containing IL-1β, TNFα and NO2 − were collected and stored at −80 °C until assay.
IL-1β assay
The measurement of IL-1β activity in different samples was conducted by thymocyte proliferation assay as described previously (Wang et al. 2002). IL-1β activity was determined by comparison with the recombinant human IL-1β (rhIL-1β) curves of serial dilutions (Palmer et al. 2003).
TNFα assay
TNFα activity in different samples was measured by using the cytotoxicity assay against L929 cells as described previously (Wang and Chen 1998). One unit of TNFα (equivalent to 25 ng/L of recombinant human TNFα) was defined as the reciprocal of the dilution producing 50 % maximal killing of L929 cells.
Nitrite (NO2 −) assay
NO production was evaluated by measuring NO2 − content in culture supernatants described previously (Wang et al. 2004). NO2 − content was measured by adding 100 μl of freshly prepared Griess reagent (equal volume of 0.2 % naphthylethylenediamine and 2 % sulfanilamide in 5 % phosphoric acid) to 100 μl of samples in 96-well plates and reading the optical density (OD) at 540 nm. The concentration of nitrite was determined by comparison with the OD curves of serial dilutions of sodium nitrite.
Examination of proteoglycan synthesis
Rabbit cartilages were isolated as described previously (Thirion and Berenbaum 2004; Wang et al. 1998). New Zealand white rabbits were sacrificed, and hind legs were prepared using routine aseptic techniques. The articular cartilages of the knee joints were removed and cut into 3 mm approximately in length. Cartilage pieces per well (30–40 mg) were added into 24-well plate with 1 ml of DMEM medium supplemented with 20 % newborn calf serum. The cultures were then incubated in the presence or absence of hrIL-1β (5 ng/ml) and/or AST (1 μM) at 37 °C in an atmosphere of 5 % CO2 for 16 h. Proteoglycan synthesis was measured by the incorporation of Na2 35SO4 (40 μCi/well) into glycosaminoglycan for additional 2 h. The 35S labeled cartilage tissues were extracted with 1 ml NaOH (0.5 M) for 48 h at room temperature and 0.5 ml aliquots applied to Sephadex G-25 P-10 columns. The eluent was counted for radioactivity. The results were expressed as cpm per mg tissue.
Chondrocyte isolation and proliferation assay
Rabbit cartilages were isolated and digested with 0.1 % hyaluronidase for 15 min followed by 0.25 % trypsin for 30 min and 0.2 % collagenase for 3 h (Thirion and Berenbaum 2004). The digested cartilage media were passed through stainless steel mesh to obtain single cell suspensions. The chondrocytes were distributed into 96-well plates (104 cells/well) in a total volume of 200 μl of DMEM medium containing 10 % newborn calf serum, penicillin (100 units/ml), streptomycin (100 μg/ml), and glutamine (2 mM) in the presence or absence of hrIL-1β (5 ng/ml) and/or AST (1 μM) for 12 h. The cultures were pulsed with 3H-TdR (1 μCi/well) for addition 6 h. The cells were digested with 0.05 % trypsin and 0.02 % ethylenediaminetetraacetic acid (EDTA) for 10 min at room temperature, and harvested on a glass fiber paper. Total radioactivity was quantified by liquid scintillation counting.
Statistical analysis
Data are presented as the mean ± SD. Comparison between groups was assessed by two-way analysis of variance (ANOVA) followed by Bonferroni’s test (Prism; GraphPad). A p value of 0.05 or less was considered statistically significant.
Results
AST suppressed joint inflammation in rat adjuvant-induced arthritis
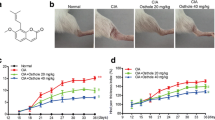
Rat adjuvant-induced arthritis (AIA) is an experimental immunopathologic model that is thought to share many features of RA (Bendele 2001). After the rats were immunized by injection of Freund’s complete adjuvant into the left hind foot pad, the animals developed the primary and acute inflammation in the joint of injected side during 1–5 days (Fig. 2a). The secondary and chronic inflammation occurred in the contralateral (non-injected) legs and became progressively severe during 7–28 days with peak response on day 21 (Fig. 2b). Recent findings show that the inflamed synovium is a major source of nitric oxide (NO) production in RA patients, and blockade of TNFα inhibits NO synthase in human peripheral blood mononuclear cells (Nagy et al. 2010; Schett et al. 2011). Treatment with NO synthase inhibitor suppresses the AIA inflammation (Stefanovic-Racic et al. 1994; Prati et al. 2012). We first tested if AST suppressed the development of AIA and NO production was involved in the effect of AST. The arthritic rats were randomly divided into AIA group, AIA treated with NO synthase inhibitor Nω-nitro-l-arginine methyl ester (l-NAME) (180 mg/kg/day), l-NAME with l-arginine (540 mg/kg/day), AST (100 mg/kg/day), and AST with l-arginine, respectively. Untreated AIA rats received an equal volume of vehicle daily. Both AST and l-NAME administrations for five consecutive days significantly inhibited primary inflammation in AIA rats (Fig. 2a). l-arginine, which is a substrate of NO synthase and inhibits the specific activity of NO synthase inhibitor, completely suppressed l-NAME effect on the paw swelling, but partially blocked AST effect on joint inflammation (Fig. 2a), suggesting that AST is not identical to NO synthase inhibitor. We then examined the effects of AST and l-NAME on the secondary inflammation in AIA rats. The data in Fig. 2b showed that both AST and l-NAME treatments for 28 days significantly suppressed the development of the chronic inflammation in AIA rats. Taken together, AST profoundly reduced both acute and chronic inflammation in AIA rats.

AST treatment inhibited primary and secondary inflammation in adjuvant-induced arthritis (AIA) rats. Rat AIA was induced by injection of 0.1 ml of Freund’s complete adjuvant into left hind foot pad on day 0. The animals were orally treated with vehicle, AST (100 mg/kg/day), l-NAME (180 mg/kg/day) and/or l-arginine (540 mg/kg/day) for different times indicated. The magnitude of the inflammatory response of injected hind paw during 1–5 days (a) and non-injected hind paw during 7–28 days (b) in each rat was evaluated described in “Materials and methods” section. Data are summarized as the mean ± SD, n = 8. *p < 0.05; **p < 0.01, compared with AIA control, AST treatment control or l-NAME treatment control
AST targeted the macrophages to modulate splenocyte proliferation in AIA rats
Many different cell components are involved in the RA development (Davignon et al. 2013; Jovanovic et al. 2011). Our data showed that the proliferation of splenocytes from AIA rats was reduced (Fig. 3a), consistent with previous reports (Albina et al. 1991; Wang and Chen 1998). Splenocytes consist of nonadherent lymphocytes and adherent monocytes/macrophages. In order to identify which cell type was AST-targeted cell, we isolated splenocytes, peritoneal macrophages and nonadherent lymphocytes (macrophages-depleted splenocytes) from normal rats, AIA rats and AIA rats treated with AST (100 mg/kg/day for 21 days). We then reconstituted cell cocultures with nonadherent splenocytes (5 × 106/ml) and peritoneal macrophages (1.25 × 105/ml). The lymphocyte proliferation in the cocultures was examined when concanavalin A (3 μg/ml) was used as the mitogenic stimulus to induce T lymphocyte proliferation (Wang and Chen 1998). The addition of macrophages from normal (Nor) rats to nonadherent lymphocytes (Lym) from AIA rats failed to reduce lymphocyte proliferation compared with both macrophages and lymphocytes from normal rats (Fig. 3b). However, addition of macrophages from AIA rats to nonadherent lymphocytes from normal or AIA rats suppressed the lymphocyte proliferation (Fig. 3b). Reconstituted cell cocultures with macrophages from AIA rats treated with AST and nonadherent lymphocytes from AIA rats restored the reduced lymphocyte proliferation, whereas addition of macrophages from AIA rats to nonadherent lymphocytes from AIA rats treated with AST failed to reverse the lymphocyte proliferation (Fig. 3b). Taken together, these data demonstrated that macrophage in splenocytes mediated the reduced lymphocyte proliferation in AIA rats. AST treatment restored the reduced lymphocyte proliferation in AIA rats through the modulation of macrophage function (Fig. 3).

AST targeted the macrophages and restored the reduced splenocyte proliferation in AIA rats. Rat AIA were induced as described in Fig. 2. The normal rats or AIA rats were treated with vehicle or AST (100 mg/kg/day) for 21 days. On day 22, the peritoneal macrophages, splenocytes and nonadherent lymphocytes in each group (five animals) were prepared as described in “Materials and methods” section. The splenocyte proliferation in coculture of nonadherent lymphocytes (5 × 106/ml) with adherent macrophages (1.25 × 105/ml) from the animal in the presence of concanavalin A (3 μg/ml) was measured by [3H]-labeled thymidine incorporation. Data are summarized as the mean ± SD, n = 5. **p < 0.01, compared with normal control. Nor normal, Mφ macrophages, Lym lymphocytes
AST suppressed IL-1β, TNFα, and NO formation in macrophages from AIA rats
Macrophages play a pivotal role in RA pathogenesis because they largely locate in the inflamed synovium and at the cartilage-pannus junction (Kinne et al. 2000). The inflammatory mediators secreted by macrophages cause pathological changes in RA patients and AIA rats (Kinne et al. 2000; Bendele 2001). We asked if the suppression of rat AIA development by AST was involved in the inhibition of inflammatory mediators produced by macrophages. AIA rats were treated with vehicle or AST (100 mg/kg/day) for 21 days. A group of untreated non-arthritic and age-matched rats was used as normal control. After termination of AST or vehicle treatment, the rats were sacrificed and peritoneal macrophages were prepared. The IL-1β, TNFα and NO productions in different groups were measured. The results in Fig. 4 showed that there were dramatic increases of IL-1β, TNFα and NO formation in macrophages from AIA rats compared with that from normal rats, which were consistent with previous reports that IL-1β, TNFα and NO participated in the pathogenesis of RA (Nagy et al. 2010; Jouzeau et al. 1999). AST treatment markedly inhibited the production of IL-1β, TNFα and NO in macrophages from AIA rats.

AST suppressed IL-1β, TNFα, and NO formation in macrophages from AIA rats. AIA rats were induced as described in Fig. 2. The normal or AIA rats were treated with vehicle or AST (100 mg/kg/day) for 21 days. On day 22, the peritoneal macrophages were cultured with lipopolysaccharide (10 μg/ml) in each group. IL-1β, TNFα and NO formation were measured as described in “Materials and methods” section. Data are summarized as the mean ± SD, n = 5. **p < 0.01, compared with normal control
AST inhibited IL-1β-induced knee joint swelling in rats
IL-1β is a potent inflammatory cytokine involved in many important cellular functions including the induction of synovitis (Presle et al. 1999). To investigate the effect of AST on IL-1β-induced joint inflammation, we intraarticularly injected recombinant human IL-1β (rhIL-1β) into the rat knee joint. The inflammation of knee joint was monitored by assessment of joint swelling. The data in Fig. 5 showed that the knee joints of animals given three injections of rhIL-1β exhibited a significant increase in joint volume as a consequence of edema, which was consistent with previous reports (Presle et al. 1999; Jouzeau et al. 1999) that IL-1β articular injection directly caused joint inflammation. AST oral administration does-dependently inhibited rhIL-1β-induced knee joint swelling (Fig. 5).

AST inhibited IL-1β-induced knee joint swelling in rats. The rats were given 50 μl rhIL-1β by intraarticular injection to the right hind knee joint (1 μg/knee) on days 0, 2 and 4. An equal volume of sterile physiological saline was injected into the left knee as a control. AST (100 mg/kg/day) or vehicle were orally treated for five consecutive days. At the end of the experiment (day 5), the animals were sacrificed and the volume of both knee joints in each rat was measured. The difference of knee joint volume in each rat was expressed as knee joint swelling. Data are summarized as the mean ± SD, n = 6. **p < 0.01, compared with control
AST in vitro concentration-dependently inhibited IL-1β production in macrophages
AST suppressed the inflammation in AIA rats and inhibited the macrophage activation. We then examined the AST effects in vitro on IL-1β production in peritoneal macrophages from normal rats. Our data showed that AST in the range of (10−10–10−6 M) concentration-dependently inhibited IL-1β formation in macrophages in the presence of lipopolysaccharide (10 μg/ml), which induced macrophage activation (Fig. 6a). The IC50 of AST was 0.16 μM. The toxicity of AST on macrophages was measured by colorimetric MTT assay, and higher concentrations of AST had no toxicity on macrophages (Fig. 6b). AST exhibited similar effects on the production of TNFα and NO in macrophages (data not shown). Together, these data demonstrated that higher concentrations of AST inhibited IL-1β production was not due to its toxicity.

AST concentration-dependently inhibited IL-1β production in macrophages in vitro. Rat peritoneal macrophages were prepared and cultured with AST(10−10 to 10−6 M) in the presence of lipopolysaccharide (10 μg/ml) for 48 h. The effects of different concentrations of AST on IL-1β production (a) and on the toxicity of macrophages (b) were measured described in “Materials and methods” section. Data are summarized as the mean ± SD of 5 independent experiments, n = 5. *p < 0.05; **p < 0.01, compared with vehicle control
AST protected against IL-1β-suppressed chondrocyte proliferation and cartilage proteoglycan synthesis in vitro
A large amounts IL-1β was found within the localized joints of RA patients, which was thought to play a key role in cartilage damage (Presle et al. 1999). IL-1β in vitro directly inhibits cartilage proteoglycan synthesis and chondrocyte proliferation (Jouzeau et al. 1999). We investigated the effects of AST in vitro on chondrocyte proliferation. The result in Fig. 7a, b showed that rhIL-1β (5 ng/ml) significantly suppressed rabbit chondrocyte proliferation and increased NO production. AST (1 μM) protected against rhIL-1β-induced the impairment of the chondrocyte proliferation by inhibiting NO production. We finally observed the AST in vitro on rhIL-1β-suppressed cartilage proteoglycan synthesis. Again, rhIL-1β significantly increased NO formation and suppressed rabbit proteoglycan synthesis (Fig. 7c, d). AST (1 μM) prevented rhIL-1β-induced damage of cartilage proteoglycan synthesis by inhibiting NO formation in chondrocytes. Taken together, our data demonstrated that AST regulated the production and activity of IL-1β, and the antiarthritic effect of AST was correlated to its preventing the inflammatory mediator-caused cartilage destruction.

AST protected against IL-1β-suppressed chondrocyte proliferation and cartilage proteoglycan synthesis in vitro. The rabbit cartilages and chondrocytes were prepared and incubated with vehicle, AST (1 μM) or hrIL-1β (5 ng/ml). The chondrocyte proliferation and NO formation (a and b) and cartilage proteoglycan synthesis and NO production (c and d) were measured described in “Materials and methods” section. Data represent the mean ± SD of 4–5 independent experiments. **p < 0.01, compared with vehicle control
Discussion
The inflamed synovial tissue in RA is characterized by a complex interplay between multiple immune cells and inflammatory mediators (Davignon et al. 2013; Zivojinovic et al. 2012). IL-17 and Th17-associated cytokines could be crucial in the pathogenesis of RA. Monocytes/macrophages from inflamed joints have been shown to induce the development of Th17, thus contributing to the amplification of inflammation (Evans et al. 2009). Recent studies showed that AST possessed immunomodulating effects on lymphocyte proliferation and proinflammatory cytokine production (Cho and Leung 2007; Wang et al. 2002). We therefore investigated the antiarthritic effects of AST in vivo and in vitro. The effects of AST on the primary and secondary inflammation were explored in AIA rats. While we examined the antiarthritic effects of AST, the similarity and difference with NO synthase inhibitor were also compared. We found both NO synthase inhibitor l-NAME and AST suppressed the inflammation in AIA rats (Fig. 2). l-Arginine completely reversed the effect of NO synthase inhibitor, but partially blocked AST effect on inhibiting joint inflammation. These findings suggest that AST is not identical to NO synthase inhibitor, and it has other effects on modulating inflammatory cell activation and response.
The defects in suppressor T cell function and reduced splenocyte proliferation are the features of immune disorders of RA and AIA (Davignon et al. 2013; Kinne et al. 2000). Splenocytes contain both lymphocytes and macrophages. In order to identify which cell type is AST targeting cell, we isolated the lymphocytes from spleen by collecting the nonadherent splenocytes and the peritoneal macrophages from peritoneal lavage by collecting the adhesive cells. We then reconstituted the co-culture of lymphocytes and macrophages. We found that coculture of lymphocytes from AIA rats with macrophages from normal rats failed to reduce the splenocyte proliferation, while assembly of lymphocytes from normal rats with macrophages from AIA rats did inhibit the splenocyte proliferation (Fig. 3). The macrophages from AIA rats treated with AST could reverse the proliferation of lymphocytes from AIA rats (Fig. 3). We also found that IL-1β, TNFα and NO production in macrophages from AIA rats were significantly higher than that from normal rats. Treatment of AST markedly inhibited the production of these inflammatory mediators in macrophages from AIA rats (Fig. 4). Therefore, AST restored the reduced splenocyte proliferation, which was mediated by macrophage activation in AIA rats. These data not only confirmed previous reported that macrophage activation caused the reduced the splenocyte proliferation but also identified that macrophage was one of AST targeted cell. Further studies are needed to determine the effects of AST on the function of synoviocytes and osteoclasts, which are involved in the RA development.
Notably, more fold-stimulation of IL-1β formation was found in comparison with that of TNFα and NO production in AIA rats (Fig. 4). IL-1β in the localized joints is thought to play a key role in cartilage damage by suppressing the synthesis of the extracellular matrix components and by promoting their degradation through metalloproteinases (Presle et al. 1999). We then observed the effect of articular injection of IL-1β to rat knee joint and found that IL-1β induced the joint swelling (Fig. 5). AST treatment suppressed IL-1β-induced knee joint edema in rats. Recent data showed that IL-1β in vitro also stimulated NO production in chondrocytes and caused the cartilage damage (Jouzeau et al. 1999; Stefanovic-Racic et al. 1997). We performed the experiments of in vitro IL-1β effect on cartilage destruction in the presence or absence of AST. IL-1β inhibited both chondrocyte proliferation and cartilage proteoglycan synthesis in vitro and increased NO formation (Fig. 7). We found AST protected against IL-1β-induced cartilage degradation by inhibiting NO production. Th17/Treg balance controls inflammation of experimental arthritis (Deng et al. 2010; Yao et al. 2011). The antiarthritic effects of AST on the modulation of Th17/Treg balance in animal model will be investigated in the future studies.
In summary, macrophages are one of AST targeted cells. In addition to their central role in inflammation, macrophages are at the origin of pathological bone erosion in RA due to their excessive differentiation into osteoclasts, which are the only cells specialized in bone resorption (Davignon et al. 2013; Takayanagi 2007). Macrophage activation and inflammatory response play an important role in mediating the joint inflammation and damage in AIA rats. AST possesses antiarthritic effects by inhibiting macrophage activation and preventing inflammatory mediator-elicited cartilage and bone destruction. These findings provide an important insight that AST may be a candidate for the treatment of RA and other inflammatory joint diseases.
References
Albina, J.E., J.A. Abate, and W.L. Henry Jr. 1991. Nitric oxide production is required for murine resident peritoneal macrophages to suppress mitogen-stimulated T cell proliferation. Role of IFN-gamma in the induction of the nitric oxide-synthesizing pathway. Journal of Immunology 147: 144–148.
Bao, F., P. Wu, N. Xiao, F. Qiu, and Q.P. Zeng. 2012. Nitric oxide-driven hypoxia initiates synovial angiogenesis, hyperplasia and inflammatory lesions in mice. PLoS One 7: e34494.
Bendele, A. 2001. Animal models of rheumatoid arthritis. Journal of Musculoskeletal and Neuronal Interactions 1: 377–385.
Chen, D.Y., Y.M. Chen, H.H. Chen, C.W. Hsieh, C.C. Lin, and J.L. Lan. 2011. Increasing levels of circulating Th17 cells and interleukin-17 in rheumatoid arthritis patients with an inadequate response to anti-TNF-alpha therapy. Arthritis Research & Therapy 13: R126.
Cho, W.C., and K.N. Leung. 2007. In vitro and in vivo immunomodulating and immunorestorative effects of Astragalus membranaceus. Journal of Ethnopharmacology 113: 132–141.
Davignon, J.L., M. Hayder, M. Baron, J.F. Boyer, A. Constantin, F. Apparailly, R. Poupot, and A. Cantagrel. 2013. Targeting monocytes/macrophages in the treatment of rheumatoid arthritis. Rheumatology (Oxford) 52: 590–598.
Deknuydt, F., G. Bioley, D. Valmori, and M. Ayyoub. 2009. IL-1beta and IL-2 convert human Treg into T(H)17 cells. Clinical Immunology 131: 298–307.
Deng, S., Y. Xi, H. Wang, J. Hao, X. Niu, W. Li, Y. Tao, and G. Chen. 2010. Regulatory effect of vasoactive intestinal peptide on the balance of Treg and Th17 in collagen-induced arthritis. Cellular Immunology 265: 105–110.
Evans, H.G., N.J. Gullick, S. Kelly, C. Pitzalis, G.M. Lord, B.W. Kirkham, and L.S. Taams. 2009. In vivo activated monocytes from the site of inflammation in humans specifically promote Th17 responses. Proceedings of the National Academy of Sciences of the United States of America 106: 6232–6237.
Hu, W., L.J. Xia, F.H. Chen, F.R. Wu, J. Tang, C.Z. Chen, S. Jiang, and H.H. Chen. 2012. Recombinant human endostatin inhibits adjuvant arthritis by down-regulating VEGF expression and suppression of TNF-alpha, IL-1beta production. Inflammation Research 61: 827–835.
Jiang, J.B., J.D. Qiu, L.H. Yang, J.P. He, G.W. Smith, and H.Q. Li. 2010. Therapeutic effects of astragalus polysaccharides on inflammation and synovial apoptosis in rats with adjuvant-induced arthritis. International Journal of Rheumatic Diseases 13: 396–405.
Jouzeau, J.Y., C. Cipolletta, N. Presle, P. Netter, and B. Terlain. 1999. Modulation of IL-1 effects on cartilage by NO synthase inhibitors: pharmacological studies in rats. Osteoarthritis Cartilage 7: 382–385.
Jovanovic, D.V., L. Boumsell, A. Bensussan, X. Chevalier, A. Mancini, and J.A. Di Battista. 2011. CD101 expression and function in normal and rheumatoid arthritis-affected human T cells and monocytes/macrophages. Journal of Rheumatology 38: 419–428.
Ju, J.H., Y.J. Heo, M.L. Cho, J.Y. Jhun, J.S. Park, S.Y. Lee, H.J. Oh, S.J. Moon, S.K. Kwok, K.S. Park, S.H. Park, and H.Y. Kim. 2012. Modulation of STAT-3 in rheumatoid synovial T cells suppresses Th17 differentiation and increases the proportion of Treg cells. Arthritis and Rheumatism 64: 3543–3552.
Kinne, R.W., R. Brauer, B. Stuhlmuller, E. Palombo-Kinne, and G.R. Burmester. 2000. Macrophages in rheumatoid arthritis. Arthritis Research 2: 189–202.
Lei, H., B. Wang, W.P. Li, Y. Yang, A.W. Zhou, and M.Z. Chen. 2003. Anti-aging effect of astragalosides and its mechanism of action. Acta Pharmacologica Sinica 24: 230–234.
Li, H.Q., J.D. Qiu, and L.H. Yang. 2009. Regulation of Astragalus heteropolysaccharides on synoviocytes apoptosis and proinflammatory cytokine secretion of rats with adjuvant arthritis. Yao Xue Xue Bao 44: 731–736.
Liu, Q.Y., Y.M. Yao, S.W. Zhang, and Z.Y. Sheng. 2011. Astragalus polysaccharides regulate T cell-mediated immunity via CD11c(high)CD45RB(low) DCs in vitro. Journal of Ethnopharmacology 136: 457–464.
Ma, X.Q., Q. Shi, J.A. Duan, T.T. Dong, and K.W. Tsim. 2002. Chemical analysis of Radix Astragali (Huangqi) in China: a comparison with its adulterants and seasonal variations. Journal of Agriculture and Food Chemistry 50: 4861–4866.
Nagy, G., A. Koncz, T. Telarico, D. Fernandez, B. Ersek, E. Buzas, and A. Perl. 2010. Central role of nitric oxide in the pathogenesis of rheumatoid arthritis and systemic lupus erythematosus. Arthritis Research & Therapy 12: 210.
Nistala, K., and L.R. Wedderburn. 2009. Th17 and regulatory T cells: rebalancing pro- and anti-inflammatory forces in autoimmune arthritis. Rheumatology (Oxford) 48: 602–606.
Palmer, G., D. Talabot-Ayer, I. Szalay-Quinodoz, M. Maret, W.P. Arend, and C. Gabay. 2003. Mice transgenic for intracellular interleukin-1 receptor antagonist type 1 are protected from collagen-induced arthritis. European Journal of Immunology 33: 434–440.
Prati, C., A. Berthelot, B. Kantelip, D. Wendling, and C. Demougeot. 2012. Treatment with the arginase inhibitor Nw-hydroxy-nor-l-arginine restores endothelial function in rat adjuvant-induced arthritis. Arthritis Research & Therapy 14: R130.
Presle, N., C. Cipolletta, J.Y. Jouzeau, A. Abid, P. Netter, and B. Terlain. 1999. Cartilage protection by nitric oxide synthase inhibitors after intraarticular injection of interleukin-1beta in rats. Arthritis and Rheumatism 42: 2094–2102.
Schett, G., L.C. Coates, Z.R. Ash, S. Finzel, S. Finzel, and P.G. Conaghan. 2011. Structural damage in rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis: traditional views, novel insights gained from TNF blockade, and concepts for the future. Arthritis Research & Therapy 13(Suppl 1): S4.
Stefanovic-Racic, M., K. Meyers, C. Meschter, J.W. Coffey, R.A. Hoffman, and C.H. Evans. 1994. N-monomethyl arginine, an inhibitor of nitric oxide synthase, suppresses the development of adjuvant arthritis in rats. Arthritis and Rheumatism 37: 1062–1069.
Stefanovic-Racic, M., M.O. Mollers, L.A. Miller, and C.H. Evans. 1997. Nitric oxide and proteoglycan turnover in rabbit articular cartilage. Journal of Orthopaedic Research 15: 442–449.
Takayanagi, H. 2007. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nature Reviews Immunology 7: 292–304.
Thirion, S., and F. Berenbaum. 2004. Culture and phenotyping of chondrocytes in primary culture. Methods in Molecular Medicine 100: 1–14.
Van Bezooijen, R.L., L. Van Der Wee-Pals, S.E. Papapoulos, and C.W. Lowik. 2002. Interleukin 17 synergises with tumour necrosis factor alpha to induce cartilage destruction in vitro. Annals of the Rheumatic Diseases 61: 870–876.
Walter, G.J., H.G. Evans, B. Menon, N.J. Gullick, B.W. Kirkham, A.P. Cope, F. Geissmann, and L.S. Taams. 2013. Interaction with activated monocytes enhances cytokine expression and suppressive activity of human CD4 + CD45ro + CD25 + CD127(low) regulatory T cells. Arthritis and Rheumatism 65: 627–638.
Wang, B., and M.Z. Chen. 1998. Effects of indomethacin on secretory function of synoviocytes from adjuvant arthritis rats. International Journal of Tissue Reactions 20: 91–94.
Wang, B., L. Ma, X. Tao, and P.E. Lipsky. 2004. Triptolide, an active component of the Chinese herbal remedy Tripterygium wilfordii Hook F, inhibits production of nitric oxide by decreasing inducible nitric oxide synthase gene transcription. Arthritis and Rheumatism 50: 303–2995.
Wang, B., Y.Y. Yao, and M.Z. Chen. 1998. Effects of indomethacin on joint damage in rat and rabbit. Zhongguo Yao Li Xue Bao 19: 70–73.
Wang, Y.P., X.Y. Li, C.Q. Song, and Z.B. Hu. 2002. Effect of astragaloside IV on T, B lymphocyte proliferation and peritoneal macrophage function in mice. Acta Pharmacologica Sinica 23: 263–266.
Yang, L.H., J.D. Qiu, and H.Q. Li. 2009. Effects of Astragalus heteropolysaccharides on erythrocyte immune adherence function of mice with adjuvant-induced arthritis. Yao Xue Xue Bao 44: 1364–1370.
Yao, R., Y. Fu, S. Li, L. Tu, X. Zeng, and N. Kuang. 2011. Regulatory effect of daphnetin, a coumarin extracted from Daphne odora, on the balance of Treg and Th17 in collagen-induced arthritis. European Journal of Pharmacology 670: 286–294.
Yu, Q.T., L.W. Qi, P. Li, L. Yi, J. Zhao, and Z. Bi. 2007. Determination of seventeen main flavonoids and saponins in the medicinal plant Huang-qi (Radix astragali) by HPLC-DAD-ELSD. Journal of Separation Science 30: 1292–1299.
Zivojinovic, S.M., N.N. Pejnovic, M.N. Sefik-Bukilica, L.V. Kovacevic, Ii Soldatovic, and N.S. Damjanov. 2012. Tumor necrosis factor blockade differentially affects innate inflammatory and Th17 cytokines in rheumatoid arthritis. Journal of Rheumatology 39: 18–21.
Acknowledgments
We would like to thank Wei-Ping Li and Ai-Wu Zhou for their excellent technical assistances.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, B., Chen, MZ. Astragaloside IV possesses antiarthritic effect by preventing interleukin 1β-induced joint inflammation and cartilage damage. Arch. Pharm. Res. 37, 793–802 (2014). https://doi.org/10.1007/s12272-014-0336-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-014-0336-2