
Abstract
Peripartum cardiomyopathy (PPCM) is an uncommon complication of pregnancy. Early case reports identified overlap between familial dilated cardiomyopathy (DCM) and PPCM, although the degree of overlap is largely unknown. Other evidence supporting a contribution from gene mutations in PPCM includes familial occurrence, genome-wide association studies, variable prevalence among different regions and ethnicities, and more recent investigations of panels of genes for mutations among women with PPCM. Murine models implicate the role of altered metabolism and increased free radical stress to the heart during pregnancy, which seems to be involved in the pathogenesis of this condition. Although the true incidence of genetic cardiomyopathy is not yet known among women with PPCM, there is substantial evidence demonstrating that at least 10–15% of affected women have a clear genetic contribution to their condition. With this in mind, family counseling, cascade phenotypic screening, and clinical genetic testing should be considered among women with PPCM.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Peripartum cardiomyopathy (PPCM) was first described in the mid-nineteenth century as a “postpartal disease,” establishing an association between acute heart failure and the puerperium [1,2,3]. But, it was only in 1937 when Gouley et al. [4] described the clinical and pathological features of seven pregnant patients that it was first recognized as a distinct clinical entity [5]. These women were described as having dilated cardiomyopathy without any ischemic disease toward the end of their pregnancies, with persistence after delivery. Four of the seven patients died soon after their diagnosis, and autopsies showed enlarged hearts with widespread severe focal areas of necrosis and fibrosis. Many authors have subsequently reported patients with heart failure around the time of delivery.
In 1971, Demakis et al. described a case series of 27 women with pregnancy-associated cardiomyopathy and introduced the term “peripartum cardiomyopathy” [6]. These authors established its diagnostic criteria as follows: (1) the development of heart failure in the last month of pregnancy or within 5 months of delivery and (2) the absence of preexisting cardiac dysfunction or any other determinable cause of cardiomyopathy. This definition of PPCM was updated in 2000 following a workshop held by the National Heart, Lung, and Blood Institute (NHLBI) and the Office of Rare Disease of the National Institutes of Health (NIH), to include echocardiographic evidence of left ventricular systolic dysfunction (left ventricular ejection fraction, LVEF < 45%, fractional shortening of < 30%, or both), with or without a left ventricular end-diastolic dimension of > 2.7 cm/m2 body surface area [7].
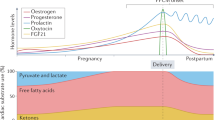
Over the years, epidemiologic data show that the incidence of PPCM is much higher in Haiti, Nigeria, and South Africa, and also among African Americans in the USA [8]. In studying whether African descent should be deemed as a risk factor for PPCM, one study compared PPCM populations in Haiti and in Martinique, an island of the French West Indies, as both countries are comprised of mostly African descendants [9]. The incidence of PPCM reported in Martinique turned out to be more than tenfold lower than that in Haiti, but this is insufficient to form conclusions, due to inherent discrepancies between the two populations, such as the differences in socioeconomic status. More research should be done to look into whether race or geographical location is an independent risk factor for PPCM (Fig. 1).

Summary of potential contributing risk factors for peripartum cardiomyopathy. MnSOD manganese superoxide dismutase, PRL prolactin, PVR peripheral vascular resistance, ROS reactive oxygen species
Increased blood volume, heart rate, and demand for higher cardiac output accompany normal pregnancies. Accordingly, women with preexisting but asymptomatic dilated cardiomyopathy (DCM) may present with new symptoms or a new diagnosis during pregnancy, although this presentation will typically occur with evidence of prior DCM, such as LV dilation and severe LV dysfunction. [7]. A small group of women, who are found to have new heart failure symptoms with new LV systolic dysfunction prior to the last gestational month, are diagnosed with pregnancy-associated cardiomyopathy (PACM) [10]. In one study, Elkayam et al. noted that comparison between PPCM and PACM groups did not show any differences in age, race, obstetric or gestational hypertension history, LVEF at diagnosis, and rate and time of recovery as well as maternal outcome. Given such findings, the authors suggested that PPCM and PACM are in fact a representation of a spectrum of the same disease.
That aside, the current diagnostic criteria for PPCM are insufficient to separate those who truly have PPCM/PACM from those who may have some underlying undiagnosed cardiac disease, which is then aggravated by pregnancy. Many of the profound hemodynamic changes in the cardiovascular system during pregnancy include increased heart rate, decreased afterload via peripheral arterial vasodilation, increased preload from increased blood volume, increased stroke volume, and increased cardiac output. Prior reviews provide a good summary of the changes in maternal cardiac metabolism during pregnancy [11] and elucidate some core differences between pregnancy-induced cardiac hypertrophy and that which is exercise-induced [12]. The latter also compared differences between physiological and pathological changes. While both articles provide substantial information in this area, there are still a lot of unresolved questions in pregnancy-related cardiac metabolism. It is thus imperative that we improve our understanding of normal metabolic changes in pregnancy, in order to study the pathogenesis of PPCM. Only then can we better discern “true PPCM” from a preexisting, undiagnosed cardiomyopathy. By doing so, we would be able to better inform patients with PPCM with regard to family planning and prognosis and provide appropriate counseling of their respective conditions. Current treatment modalities for PPCM are similar to other cardiomyopathies, but better understanding of the condition may give rise to a more effective and targeted novel therapy in the future.
To date, there have been many hypotheses on the causes of PPCM, including but not limited to fetal microchimerism related to the maternal autoimmune response [13], inflammation, increased oxidative stress, viral infection, imbalance of cardiac proapoptotic and antiangiogenic factors, micronutrient deficiency, and, last but not least, genetic susceptibility [14]. Because of the complexity of PPCM, genetic contributions could plausibly be anywhere on the spectrum from extremely rare variants with large effects to relatively common variants with smaller effects. The purpose of this review is to summarize the evidence for genetic susceptibility in the pathogenesis of PPCM, as well as the associated pathways involved with development of this uncommon form of cardiomyopathy.
PPCM as a Subset of Familial DCM
One of the earliest observations on the familial clustering of PPCM was described in 1963 in a retrospective case series of 17 patients over a 12-year period. Three of the 17 patients gave a definite family history of PPCM, known as “postpartal heart failure” [15]. A few years later, Hughes et al. described a fatal case of PPCM [16], followed by an extensive family history of cardiomyopathy in the next 16 years [17], not only with the occurrence of PPCM in the sister but also in two nephews who were diagnosed with cardiomyopathy at ages 12 years and younger. Other similar reports include a patient with fatal PPCM, with her mother and two of six sisters with the same fate [18]; a 16-year-old girl with biopsy-proven cardiomyopathy after a molar pregnancy, with her sister who had a cardiac transplantation for PPCM [19]; a 24-year-old woman who died of ventricular fibrillation 15 months after the delivery of her third child and diagnosis of PPCM, whose 34-year-old mother died of cardiomyopathy 1 week after the delivery of her fifth child, and whose sister was also diagnosed with heart failure 2 days after the delivery of her second child and died from heart failure at 26 years of age [20].
These examples of familial clustering initially implicated a genetic basis for PPCM, but they could not adequately discern atypical examples from a more common substrate in the pathogenesis of PPCM. With those leads, van Spaendonck-Zwarts et al. reviewed their database of DCM for the presence of PPCM cases, followed by a review of their PPCM cases for any family members with undiagnosed DCM [21]. In the first part of their study, they sent out invitation letters to family members of patients with idiopathic DCM, asking if they would agree to presymptomatic cardiologic screening, with a specific intention of identifying PPCM cases. DCM is diagnosed when a patient has both reduced LVEF of < 0.45 and dilation of the left ventricle with LVEDd of > 117% of predicted value corrected for body surface area and age, after identifiable causes like severe hypertension, coronary artery disease, and systemic diseases have been ruled out. Familial DCM is diagnosed if there are two or more affected family members, or if a first-degree relative of a DCM patient died suddenly before 35 years of age [22]. They found that, among 90 families with idiopathic DCM, five families had at least one individual with PPCM. In the second part of their study, they evaluated ten PPCM cases and found five patients who did not fully recover within a year based on the criteria for DCM. They were able to further evaluate family members in three of these five patients and found that all three families included first-degree relatives with previously unrecognized DCM. Genetic analysis of three DCM genes (LMNA, TNNT2, and MYH7) identified no mutations. They did, however, find a mutation in TNNC1 (p.Gln50Arg), which was designated as pathogenic because of the following: (1) it alters the highly conserved glutamine residue and is surrounded by conserved residues; (2) the amino acid substitution is localized in a small critical linker region known to be involved in protein-protein interactions; (3) the mutation cosegregates with disease in this family; (4) the mutation was absent in 300 alleles from ethnically matched controls; and (5) the mutation is classified as pathogenic by several prediction algorithms. This was the first systematic study that looked into the relationship between PPCM and DCM, and the results supported their hypothesis that PPCM may indeed be a subset of familial DCM. Granted, this was a small subset of their PPCM database that was analyzed. The authors acknowledged that PPCM patients who fully recovered within a year should also be screened for potential family members with undiagnosed DCM. They also recognized the need for a larger patient cohort to reassess the true incidence of gene mutations among women with PPCM.
Another similar study evaluated genetic variants in patients with PPCM [23]. Among 4110 females from 520 pedigrees in their familial DCM database, they identified 45 individuals with PPCM, 19 of whom were analyzed for mutations in 14 DCM genes. Six (13%) were found to have mutations, five of whom had PPCM. In this cohort, three had a family history of DCM, with heterozygous mutations in MYH7, SCN5A, and PSEN2; two others were “sporadic” mutations in MYH6 and TNNT2, in that no family history was available and the variants were not seen in the controls. An additional mutation was found in MYBPC3 in a PACM patient. This further supports the hypothesis that a subset of PPCM may be part of a spectrum of familial DCM, and that the hemodynamic and/or hormonal stresses of pregnancy unmask the underlying disease, causing heart failure.
In recent years, TTN has gained a lot of attention for its role in DCM and some adult-onset inherited skeletal muscle diseases, such as tibial muscular dystrophy (TMD) (MIM#600334), limb-girdle muscular dystrophy type 2J (LGMD2J) (MIM#608807), and hereditary myopathy with early respiratory failure (HMERF) (MIM#603689) [24]. TTN encodes the largest human protein, titin, and it is also the third most abundant striated-muscle protein [25]. Titin acts as the primary scaffold for the organization and assembly of the sarcomere, and it is the defining structural element that spans from the Z-disk to the M-line [24]. As it is involved in key structural, developmental, mechanical, and regulatory functions in cardiac and skeletal muscles, TTN is important for understanding the genetic causes for numerous diseases such as those noted above. Its large size (363 exons) correlates with the high likelihood of finding mutations in this gene. Yet until recently, because of its gigantic size and complexity, it has been incompletely studied. The introduction of next-generation sequencing has helped to improve genetic research on this front, allowing us to better understand the genetic variation in TTN.
One of the earliest studies of TTN included 312 probands with non-ischemic DCM, recognizing truncations in this gene in 67/312 (21%), predominantly occurring in the A-band [26]. In contrast, 7/249 (3%) controls without cardiomyopathy and 3/231 (1%) people with hypertrophic cardiomyopathy (HCM) had truncations in this gene. Each of the three people with HCM and TTN truncations also had a pathogenic mutation in either MYH7 or MYBPC3. In contrast with DCM, most of the TTN truncations for controls (without cardiomyopathy or with HCM) occurred in the N-terminal portion of this gene, typically in exons that are frequently spliced out of cardiac mRNA. Subsequent analyses confirm that truncations in TTN are the most common genetic cause of DCM [27,28,29,30].
Genetic investigation was subsequently performed for PPCM, due to the association between PPCM and familial DCM. The prevalence of TTN truncations prompted analysis for truncations of this and other genes in PPCM patients. A Dutch study applied genetic analysis of 48 genes among 18 families with both PPCM and DCM. They identified four pathogenic truncations in four of the 18 families (22%), three in TTN and one in BAG3, as well as six other variants of unknown significance (VUS) that may be pathogenic (33%) [31]. Four of the VUS were truncations in TTN that were located in the A-band, establishing a high prevalence of TTN A-band truncations in PPCM (7/18, 39%). However, this study was enriched for discovery of gene mutations, since the participants were women with a diagnosis of PPCM who also had other affected family members [31].
More recently, a study was carried out by analyzing 43 genes in 172 women with PPCM [32]. Participants were recruited from six independent cohorts, the largest of which was 83 women from the Investigations in Pregnancy Associated Cardiomyopathy (IPAC) trial, a multicenter, prospective study of women with peripartum cardiomyopathy [33]. Genetic evaluation utilized next-generation DNA sequencing, focusing on truncating variants rather than on missense alleles. In this cohort, 26/172 (15%) had a truncation identified in one of the eight truncating genes. The prevalence of truncating TTN variants in this group was compared with those in DCM patients and in population controls. The results were consistent with those of prior studies, such that prevalence of truncating TTN variants in PPCM was 10% (17/172), which was indeed higher than that in the control groups. The authors also identified heterozygous truncations in two genes on the X chromosome (DMD and LAMP2), as well as truncations in DSP, MYH6, SYNM, TPM1, and VCL. Among these genes, both MYH6 and SYNM have truncations in more than 0.5% of controls in enlarging databases of human exomes, suggesting that these were not pathogenic. Although the prevalence of truncations in these genes in controls may suggest that this analysis overestimates the incidence of genetic DCM among women with PPCM, it is important to recognize that many of the genes analyzed (22/47; 47%) typically have pathogenic missense mutations instead of truncations causing DCM. Inclusion of rare missense variants in this analysis would almost certainly have led to a conclusion of a much higher prevalence of genetic DCM among women with PPCM. Table 1 lists genes in which mutations have been reported in association with PPCM. The addition of other genes in which mutations are known to cause DCM would probably also have recognized a higher incidence of genetic DCM in PPCM.
PPCM in Female Carriers of X-Linked Forms of Cardiomyopathy
A case report in 2001 noted that a young 25-year-old woman, who presented with PPCM during her 36th week of pregnancy, required a left ventricular assist device (LVAD) after delivery due to severe left ventricular systolic dysfunction and eventually underwent a cardiac transplantation [34]. Her past medical history was significant for being a known carrier of Duchenne muscular dystrophy (DMD). During LVAD placement, the surgeons noted severe enlargement of all the cardiac chambers, which was consistent with acute exacerbation of chronic cardiomyopathy. While it is unclear whether this patient had an undiagnosed preexisting cardiomyopathy due to her carrier state for DMD, or whether she had a PPCM unrelated to her carrier status, she did meet the diagnostic criteria of PPCM in that she was well with no heart failure symptoms before her final month of pregnancy.
A cohort study in 1996 looked at the development of cardiomyopathy in female carriers of Duchenne and Becker muscular dystrophies (BMD) [35]. Results showed that there was preclinical or clinically evident myocardial involvement in 84.3% of the total cohort (166 of 197 cases), without significant differences in percentage and behavior between DMD and BMD carriers. Years later, a case report on a 40-year-old woman who was diagnosed with and treated for PPCM was found to be a carrier of DMD mutant gene after her son was diagnosed with DMD at 4 years of age [36]. Another similar case was reported about a 25-year-old primigravid woman who was diagnosed with PPCM, and who was later found to have a de novo mutation in Xp21 when her son was diagnosed with DMD [37]. The fact that dystrophinopathies can cause myocardial damage is well known, but what is yet unknown is whether there exists some non-random X-chromosome inactivation (XCI) pattern in the small number of female carriers of D/BMD with PPCM, with the caveat that the actual incidence of PPCM in DMD/BMD carriers is still unclear.
One study looked at the clinical and genetic characterization of manifesting carriers of DMD mutations [38]. While they noted five out of 15 manifesting carriers with certain mutations also had cardiomyopathy (only one of them with PPCM), they did not include asymptomatic carriers, which as seen in the two cases noted above is entirely possible to develop PPCM regardless of their DMD manifestation.
Finally, in the Ware manuscript, the two individuals who had truncating variants in DMD and LAMP2 were alluded to possibly having peripartum cardiomyopathy as a consequence of skewed X-chromosome inactivation [32, 39]. Two other reports identified individuals with PPCM who had a DMD or LAMP2 mutation, the latter of which causes Danon disease [36, 40]. This X-linked disorder presents in young males with developmental delay, skeletal myopathy, and LVH or hypertrophic cardiomyopathy. It typically progresses to severe systolic dysfunction requiring cardiac transplantation by age 20.8 ± 6.7 years in male patients, with the mean age of death at 20.1 ± 5.2 years [41]. Female carriers of Danon disease often have later onset of symptoms, but eventually develop cardiomyopathy and heart failure as well, with the mean age of cardiac transplantation at 32.3 ± 14.5 years and mean age of death at 40.2 ± 12.6 years [41]. The incidence of PPCM among females with heterozygous LAMP2 mutations is not known, due to the rare nature of Danon disease. However, this association prompts concern for the progression of heart disease among pregnant women with LAMP2 mutations.
Other Genomic Analyses in PPCM
The genetic contributions to PPCM noted above refer to rare variants with a major effect. Additionally, it is plausible that more common variants with a smaller effect contribute to this complex disorder. Horne et al. carried out the first genome-wide association study (GWAS) in PPCM in 2011 [42]. One single nucleotide polymorphism (SNP), rs258415, met the genome-wide significance for PPCM and was verified and replicated with a different set of cases and controls. This SNP is located at chromosome 12p11.22 near PTHLH. Parathyroid hormone-related protein (PTHrP), the product of PTHLH, is thought to be involved in preventing contractions until term and modulating ventricular contraction by exerting control of pacing at the sinus node [43]. It is also potentially involved in the etiology of preeclampsia. Given its inotropic effect on cardiac myocytes and its involvement in preventing uterine contraction, PTHrP may potentially play a role in PPCM, though further investigation is necessary.
More recently, a study that looked into the GNB3 c.825C>T polymorphism in a group of PPCM women in relation to its effect on myocardial recovery, alluded to the finding that TT genotype seemed to be more common among African American women, and that PPCM women with this genotype may be at a higher risk for chronic cardiomyopathy [44]. Although this study is limited by its small sample size and the absence of a validation cohort, it is a potential area for additional investigation.
Physiological Stress Related to Pregnancy
For women who carry a genetic predisposition to cardiomyopathy, there are probably other non-genetic factors inherent in pregnancy, which result in concurrent cardiac dysfunction. Physiological changes in pregnant women are complex and elaborate, one of them being the hemodynamic stresses on the mothers’ hearts that have an increased amount of work to do in order to deliver nutrients and oxygen to the fetus. Yet, most women who experience the normal physiologic changes inherent in pregnancy and the postpartum period do not develop cardiomyopathy. This section outlines the cardiac physiology of pregnancy and research studies implicating several aspects of the puerperium to the development of PPCM.
In normal pregnancy, the continuous stress from prolonged cardiac volume overload results in mild eccentric cardiac hypertrophy, whereby progesterone surges in early pregnancy seem to initiate this cascade [12]. Estrogen, on the other hand, appears to be antihypertrophic [45]. Relaxin, a polypeptide hormone produced by the corpus luteum during pregnancy and found within the vasculature, has also been thought to be associated with decreased vascular resistance and increased cardiac output [46]. Additional studies have shown that relaxin-2 increases nitric oxide bioavailability and lowers oxidative stress [47]. Early studies suggested that recombinant relaxin-2 (serelaxin) may have a therapeutic role for treatment of acute heart failure [48]. However, serelaxin does not prevent heart failure in an experimental model of PPCM, and unpublished phase III studies of this recombinant peptide in acute heart failure did not meet the primary endpoints of reduced cardiovascular death or worsening heart failure in patients with acute heart failure [49].
Regarding cardiac metabolism during pregnancy, the body generally tries to conserve nutrients for fetal development. Insulin resistance, gluconeogenesis during early pregnancy, and lipolysis during late pregnancy all work towards the goal of fetal growth until it is viable in the outer environment. Mechanisms underlying these metabolic changes remain incompletely understood, but a host of hormones such as β-human chorionic gonadotropin, followed by human placental lactogen (hPL), chorionic somatotropin, estrogen, progesterone, prolactin, cortisol, thyroid hormone, growth hormone, and insulin are all implicated in causing insulin resistance [11]. The upstream mechanisms or pathways that drive metabolic changes remain obscure, as are the mechanisms for how these changes correlate with PPCM.
An imbalance of cardiac angiogenic factors may contribute to the severity of PPCM. One well-studied transcriptional factor, known as signal transducer and activator of transcription-3 (STAT3), also an acute-phase response factor, plays a vital role in regulating numerous genes involved in cell survival, modulation of hypertrophic growth, angiogenesis, development and regeneration, energetics and metabolic reserve, antioxidative pathways, extracellular matrix composition, and inflammatory processes [50,51,52]. One of its many functions includes its cardioprotective role in reducing oxidative stress, in part by upregulation of ROS scavenging enzyme, manganese superoxide dismutase (MnSOD) [53]. In a study done by Hilfiker-Kleiner et al., it was shown that in cardiomyocyte-specific STAT3-knockout mice, the lack of MnSOD caused an increased level of free radicals that led to an increased level of cathepsin-D, an enzyme that cleaves prolactin into the smaller, antiangiogenic, and proapoptotic 16-kDa fragment (16K PRL), which in turn caused PPCM in those mice [50]. A pilot study that evaluated the use of bromocriptine, a dopamine receptor agonist as well as a prolactin inhibitor, showed promising results as a potential therapeutic option to improve left ventricular function in the PPCM population, even though the number of participants was small and results could not be considered definitive [54]. A recently completed randomized clinical trial will hopefully provide us with more evidence of the efficacy of the therapy (NCT00998556) [55].
A separate study found that 16K PRL specifically induced microRNA-146a (miR146a) in endothelial cells, which resulted in decreased metabolic activity and angiogenesis after exosomal transfer into cardiomyocytes [56]. The attenuation of angiogenesis was attributed to the downregulation of NRAS. When Stat3-knockout mice are treated with anti-miR146a, features of PPCM are ameliorated without interference with lactation. While the absence of the cardiomyocyte-specific STAT3 gene seems to play a part in causing PPCM in mice, and bromocriptine along with anti-miR146a may be of therapeutic benefit for this condition, there are no human genetic data to support this thus far. In a cohort study of 18 women with PPCM, no STAT3 gene mutations were discernible [31].
Apart from STAT3, another important regulator in cardiac metabolism is peroxisome proliferator-activated receptor (PPAR) gamma coactivator 1 (PGC1α), a transcriptional coactivator that is vital in mitochondrial biogenesis as well as for the expression and secretion of proangiogenic factors such as vascular endothelial growth factor (VEGF) [57]. PGC1α is also known to increase scavenging of reactive oxygen species (ROS) [58]. Mice with cardiac-specific knockout of PGC1α develop PPCM, and there is evidence that this protein was mediated by at least two separate proangiogenic pathways, namely via the VEGF pathway and the prolactin pathway [57]. In addition, soluble Flt1 (sFlt1), a splice variant of VEGF receptor, also an antiangiogenic kinase, is secreted by the placenta during late gestation, and sFlt1 alone is shown to cause cardiomyopathy in PGC-1α knockout mice [57]. Several studies have shown that high levels of sFlt1 are seen in pregnant women [57, 59], and a subset of women with preeclampsia has slightly higher levels of sFlt1 [60].
Preeclampsia has long been observed to be associated with PPCM. A systematic review and meta-analysis is supportive of the concept that preeclampsia and PPCM share similar pathogenesis [61]. However, a single-center prospective case-comparison study showed that PPCM associated with any hypertensive disease, including preeclampsia and those without any hypertensive disease, is different in the time of onset of heart failure, clinical characteristics, and outcomes, suggesting that perhaps there may be two different etiologies at work [62]. Because only a minority of women with preeclampsia develops PPCM, Patten et al. also hypothesized that PPCM may be caused by a “two-hit” combination, first of which is due to the imbalance of angiogenic factors, in particular those that involve prolactin, sFlt1, and VEGF. The second hit could be due to a number of other etiologies such as myocarditis, immune activation, viral infection and/or autoantibodies, and genetic predisposition [57]. In a case report, both preeclampsia and PPCM were found in a molar pregnancy with triploidy (69, XXX); the molecular analysis of the conceptus and parental DNA demonstrated an excess of paternal genomic contribution. This finding was suggestive of the role of genomic imprinting in PPCM [63]. Other studies in the past suggested that compromised maternal autoimmunity and the involvement of fetal microchimerism lead to autoimmune responses similar to those seen in allogeneic organ transplantation, causing PPCM [64].
Clinical Implications of Genetic Contributions to PPCM
The European Society of Cardiology, the American Heart Association, the American College of Cardiology, and the Heart Failure Societies of North America and Europe all recommend phenotypic screening (echocardiography) of first-degree relatives for individuals with idiopathic DCM [65,66,67]. Since PPCM often overlaps with idiopathic DCM, and because of the data demonstrating that a substantial subset of women with PPCM have a pathogenic gene mutation contributing to their cardiomyopathy, one can readily infer that echocardiographic screening of first-degree family members is warranted for people diagnosed with PPCM. Furthermore, with improvements in the cost and technology related to DNA analysis, the cost of clinical genetic testing is declining and may now be even less expensive than an echocardiogram [68]. Accordingly, if a patient with PPCM undergoes genetic testing with recognition of clearly pathogenic gene mutation to explain their cardiac dysfunction, gene-targeted screening of family members is likely more cost effective than serial echocardiograms in all first-degree relatives. Among women who are known heterozygous carriers of X-linked disorders that can cause cardiomyopathy, such as DMD, BMD, Danon disease, Barth syndrome, and X-linked Emery-Dreifuss muscular dystrophy, clinicians should be particularly aware of an apparent increase in the risk of PPCM, with consideration for echocardiographic evaluations in the later stages of pregnancy or early postpartum. Finally, discovery of a mutation predisposing to X-linked forms of cardiomyopathy among women with PPCM has immediate and direct consequences for their newborn male children, who may then be evaluated for either the mutation or its earlier and more severe phenotypic effects.
Conclusion
Several lines of evidence support a substantial role for genetic contributions to PPCM. These include case reports, small families, and larger multicenter investigations showing that at least 10–15% of women with PPCM have a pathogenic mutation. GWASs show additional genomic associations with this condition, and murine models highlight the roles of metabolism and free radical stress. Several studies have highlighted women who are carriers of dystrophin mutations presenting with PPCM. Future studies will better characterize the prevalence of pathogenic mutations contributing to this disorder and may also help to provide prognostic information for women who present with pregnancy during the peripartum period.
References
Virchow, R. Sitzing der Berliner Geburtshilflisher Gersellskhalt, cited by Porak, C. (1880). De l'influence réciproque de la grossesse et des maladies du Coeur, thesis, Paris.
Ritchie, C. (1850). Clinical contribution to the patho-diagnosis and treatment of certain chronic diseases of the heart. Edinburgh Medical Journal, 2, 2.
Porak, C. (1880). De l'influence réciproque de la grossesse et des maladies du Coeur, thesis, Paris.
Gouley, B. A., McMillan, T. M., & bellet S. (1937). Idiopathic myocardial degeneration associated with pregnancy and especially the puerperium. The American Journal of the Medical Sciences, 19, 185–199.
Abboud, J., Murad, Y., Chen-Scarabelli, C., Saravolatz, L., & Scarabelli, T. M. (2007). Peripartum cardiomyopathy: a comprehensive review. International Journal of Cardiology, 118(3), 295–303.
Demakis, J. G., Rahimtoola, S. H., Sutton, G. C., et al. (1971). Natural course of peripartum cardiomyopathy. Circulation, 44, 1053–1061.
Pearson, G. D., Veille, J. C., Rahimtoola, S., et al. (2000). Peripartum cardiomyopathy. National Heart Lung and Blood Institute and Office of Rare Diseases (National Institutes of Health) workshop recommendations and review. JAMA, 283, 1183–1188.
Sliwa, K., Fett, J., & Elkayam, U. (2000). Peripartum cardiomyopathy. Lancet, 368, 687–693.
Sebillotte, C. G., Deligny, C., Hanf, M., Santiago, R., Chevallier, J. C., Volumenie, J. L., & Arfi, S. (2010). Is African descent an independent risk factor of peripartum cardiomyopathy? International Journal of Cardiology, 145, 93–94.
Elkayam, U., Akhter, M. W., Singh, H., Khan, S., Bitar, F., Hameed, A., & Shotan, A. (2005). Pregnancy-associated cardiomyopathy: clinical characteristics and a comparison between early and late presentation. Circulation, 111, 2050–2055.
Liu, L. X., & Arany, Z. (2014). Maternal cardiac metabolism in pregnancy. Cardiovascular Research, 101, 545–553.
Chung, E., & Leinwand, L. A. (2014). Pregnancy as a cardiac stress model. Cardiovascular Research, 101, 561–570.
Ansari, A. A., Fett, J. D., Carraway, R. E., Mayne, A. E., Onlamoon, N., & Sundstrom, B. (2002). Autoimmune mechanisms as the basis for human peripartum cardiomyopathy. Clinical Reviews in Allergy and Immunology, 23, 301–324.
Ntusi, N. B., & Mayosi, B. M. (2009). Aetiology and risk factors of peripartum cardiomyopathy: a systematic review. International Journal of Cardiology, 131, 168–179.
Pierce, J. A., Price, B. O., & Joyce, J. W. (1963). Familial occurrence of postpartal heart failure. Archives of Internal Medicine, 111, 651–655.
Hughes, R. A. C., Kapur, P., Sutton, G. C., & Honey, M. (1970). A case of fatal peripartum cardiomyopathy. British Heart Journal, 32, 272–276.
Honey, M. (1986). Correspondence: a case of fatal peripartum cardiomyopathy. British Heart Journal, 55, 114.
Voss, E. G., Reddy, C. V. R., Detrano, R., Virmani, R., Zabriskie, J. B., & Fotino, M. (1984). Familial dilated cardiomyopathy. The American Journal of Cardiology, 54, 456–457.
Massad, L. S., Reiss, C. K., Mutch, D. G., & Haskel, E. J. (1993). Familial peripartum cardiomyopathy after molar pregnancy. Obstetrics and Gynecology, 81, 886–888.
Pearl, W. (1995). Familial occurrence of peripartum cardiomyopathy. American Heart Journal, 129, 421–422.
van Spaendonck-Zwarts, K. Y., van Tintelen, J. P., van Veldhuisen, D. J., van der Werf, R., Jongbloed, J. D. H., Paulus, W. J., Dooijes, D., & van den Berg, P. (2010). Peripartum cardiomyopathy as a part of familial dilated cardiomyopathy. Circulation, 121, 2169–2175.
Mestroni, L., Krajinovic, M., Severini, G. M., Pinamonti, B., Lenarda, A. D., Giacca, M., Falaschi, A., & Camerini, F. (1994). Familial dilated cardiomyopathy. British Heart Journal, 72(Supplement), S35–S41.
Morales, A., Painter, T., Li, R., Siegfried, J. D., Li, D., Norton, N., & Hershberger, R. E. (2010). Rare variant mutations in pregnancy-associated or peripartum cardiomyopathy. Circulation, 121, 2176–2182.
Gerull, B. (2015). The rapidly evolving role of titin in cardiac physiology and cardiomyopathy. The Canadian Journal of Cardiology, 31, 1351–1359.
Trinick, K., Knight, P., & Whiting, A. (1984). Purification and properties of native titin. Journal of Molecular Biology, 180, 331–356.
Herman, D. S., Lam, L., Taylor, M. R., et al. (2012). Truncations of titin causing dilated cardiomyopathy. The New England Journal of Medicine, 366, 619–628.
Akinrinade, O., Koskenvuo, J. W., & Alastalo, T.-P. (2015). Prevalence of titin truncating variants in general population. PloS One, 10(12), e0145284.
Roberts, A. M., Ware, J. S., Herman, D. S., Schafer, S., Baksi, J., Bick, A. G., Buchan, R. J., Walsh, R., et al. (2015). Integrated allelic, transcriptional and phenomic dissection of the cardiac effects of titin truncation in health and disease. Science Translational Medicine, 7(270), 270ra6.
Pugh, T. J., Kelly, M. A., Gowrisankar, S., Hynes, E., Seidman, M. A., et al. (2014). The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genetics in Medicine, 16(8), 601–608.
Haas, J., Frese, K. S., Peil, B., Kloos, W., Keller, A., Nietsch, R., Feng, Z., Muller, S., et al. (2015). Atlas of the clinical genetics of human dilated cardiomyopathy. European Heart Journal, 36, 1123–1135.
van Spaendonck-Zwarts, K. Y., Posafalvi, A., van den Berg, M. P., Hilfiker-Kleiner, D., Bollen, I. A. E., Sliwa, K., et al. (2014). Titin gene mutations are common in families with both peripartum cardiomyopathy and dilated cardiomyopathy. European Heart Journal, 35, 2165–2173.
Ware, J. S., Li, J., Mazaika, E., Yasso, C. M., DeSouza, T., Cappola, T. P., Tsai, E. J., Hilfiker-Kleiner, D., et al. (2016). Shared genetic predisposition in peripartum and dilated cardiomyopathies. The New England Journal of Medicine, 374, 233–241.
McNamara, D. M., Elkayam, U., Alharethi, R., et al. (2015). Clinical outcomes for peripartum cardiomyopathy in North America: results of the Investigations of Pregnancy Associated Cardiomyopathy (IPAC) study. Journal of the American College of Cardiology, 66, 905–914.
Davies, J. E., Winokur, T. S., Aaron, M. F., Benza, R. L., Foley, B. A., & Holman, W. L. (2001). Cardiomyopathy in a carrier of Duchenne’s muscular dystrophy. Journal of Heart and Lung Transplantation, 20(7), 781–784.
Politano, L., Nigro, V., Nigro, G., Petretta, V. R., Passamano, L., Papparella, S., Di Somma, S., & Comi, L. I. (1996). Development of cardiomyopathy in female carriers of Duchenne and Becker muscular dystrophies. JAMA, 275(17), 1335–1338.
Cheng, V. E., & Prior, D. L. (2013). Peripartum cardiomyopathy in a previously asymptomatic carrier of Duchenne muscular dystrophy. Heart, Lung & Circulation, 22, 677–681.
Ahmed, A., Spinty, S., Murday, V., Longman, C., & Khand, A. (2015). A de-novo deletion of dystrophin provoking severe ‘peripartum cardiomyopathy’: the importance of genetic testing in peripartum cardiomyopathy to uncover female carriers. International Journal of Cardiology, 203, 1084–1085.
Soltanzadeh, P., Friez, M. J., Dunn, D., von Niederhausern, A., et al. (2010). Clinical and genetic characterization of manifesting carriers of DMD mutations. Neuromuscular Disorders, 20, 499–504.
Giliberto, F., Radic, C. P., Luce, L., Ferreiro, V., de Brasi, C., & Szijan, I. (2014). Symptomatic female carriers of Duchenne muscular dystrophy (DMD): genetic and clinical characterization. Journal of Neurological Sciences, 336, 36–41.
Toib, A., Grange, D. K., Kozel, B. A., Ewald, G. A., White, F. B., & Canter, C. E. (2010). Distinct clinical and histopathological presentations of Danon cardiomyopathy in young women. Journal of the American College of Cardiology, 55, 408–410.
Boucek, D., Jirikowic, J., & Taylor, M. (2011). Natural history of Danon disease. Genetics in Medicine, 13(6), 563–568.
Horne, B. D., Rasmusson, K. D., Alharethi, R., et al. (2011). Genome-wide significance and replication of the chromosome 12p11.22 locus near the PTHLH gene for peripartum cardiomyopathy. Circulation. Cardiovascular Genetics, 4, 359–366.
Maioli, E., Fortino, V., & Pacini, A. (2004). Parathyroid hormone-related protein in preeclamspia: a linkage between maternal and fetal failures. Biology of Reproduction, 71, 1779–1784.
Sheppard, R., Hsich, E., Damp, K., Elkayam, U., et al. (2016). GNB3 C825T polymorphism and myocardial recovery in peripartum cardiomyopathy: results of multicenter Investigations of Pregnancy-Associated Cardiomyopathy Study. Circulation. Heart Failure, 9, e002683.
Van Eickels, M., Grohe, C., Cleutjens, J. P. M., Janssen, B. J., Wellens, H. J. J., & Doevendans, P. A. (2001). 17{beta}-estradiol attenuates the development of pressure-overload hypertrophy. Circulation, 104, 1419–1423.
Conrad, K. P. (2011). Maternal vasodilation in pregnancy: the emerging role of relaxin. American Journal of Physiology. Regulatory Integrative and Comparative Physiology, 301, R267–R275.
Sasser, J. M., Cunningham Jr., M. W., & Baylis, C. (2014). Serelaxin reduces oxidative stress and asymmetric dimethylarginine in angiotensin II-induced hypertension. American Journal of Physiology. Renal Physiology, 307, F1355–F1362.
Teerlink, J. R., Cotter, G., Davison, B. A., Felker, G. M., Filippatos, G., Greenberg, B. H., Ponikowski, P., Unemori, E., Voors, A. A., Adams Jr., K. F., Dorobantu, M. I., Grinfeld, L. R., Jondeau, G., Marmor, A., Masip, J., Pang, P. S., Werdan, K., Teichman, S. L., Trapani, A., Bush, C. A., Saini, R., Schumacher, C., Severin, T. M., Metra, M., & Investigators REiAHF. (2013). Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): a randomised, placebo-controlled trial. Lancet, 381, 29–39.
Nonhoff, J., Ricke-Hoch, M., Mueller, M., Stapel, B., Pfeffer, T., Kasten, M., Scherr, M., von Kaisenberg, C., Bauersachs, J., Haghikia, A., & Hilfiker-Kleiner, D. (2017). Cardiovascular Research, 113(6), 598–608.
Hilfiker-Kleiner, D., Kaminski, K., Podewski, E., Bonda, T., Schaefer, A., Sliwa, K., et al. (2007). A cathepsin D-cleaved 16 kDa form of prolactin mediates postpartum cardiomyopathy. Cell, 128, 589–600.
Kurdi, M., & Booz, G. W. (2009). JAK redux: a second look at the regulation and role of JAKs in the heart. American Journal of Physiology. Heart and Circulatory Physiology, 297(5), H1545–H1556.
Hilfiker-Kleiner, D., et al. (2004). Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circulation Research, 95(2), 187–195.
Negoro, S., et al. (2001). Activation of signal transducer and activator of transcription 3 protects cardiomyocytes from hypoxia/reoxygenation-induced oxidative stress through the upregulation of manganese superoxide dismutase. Circulation, 104(9), 979–981.
Sliwa, K., Blauwet, L., Tibazarwa, K., Libhaber, E., Smedema, J.-P., Becker, A., McMurray, J., Yamac, H., Labidi, S., Struman, I., & Hilfiker-Kleiner, D. (2010). Evaluation of bromocriptine in the treatment of acute severe peripartum cardiomyopathy: a proof-of-concept pilot study. Circulation, 121(13), 1465–1473.
Haghikia, A., Podewski, E., Berliner, D., Sonnenschein, K., Fischer, D., Angermann, C. E., Böhm, M., Röntgen, P., Bauersachs, J., Hilfiker-Kleiner, D. (2015). Rationale and design of a randomized, controlled multicentre clinical trial to evaluate the effect of bromocriptine on left ventricular function in women with peripartum cardiomyopathy. Clinical Research in Cardiology, 104(11), 911–917.
Halkein, J., Tabruyn, S. P., Ricke-Hoch, M., et al. (2013). MicroRNA-146a is a therapeutic target and biomarker for peripartum cardiomyopathy. The Journal of Clinical Investigation, 123, 2143–2154.
Patten, I. S., Rana, S., Shahul, S., Rowe, G. C., Jang, C., Liu, L., Hacker, M. R., Rhee, J. S., Mitchell, J., Mahmood, F., Hess, P., Farrell, C., et al. (2012). Cardiac angiogenic imbalance leads to peripartum cardiomyopathy. Nature, 485, 333–338.
St-Pierre, J., Drori, S., Uldry, M., Silvaggi, J. M., Rhee, J., Jager, S., Handschin, C., Zheng, K., Lin, J., Yang, W., Simon, D. K., Bachoo, R., & Spiegelman, B. M. (2006). Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell, 127, 397–408.
Venkatesha, S., et al. (2006). Soluble endoglin contributes to the pathogenesis of preeclampsia. Nature Medecine, 12, 642–649.
Wolf, M., et al. (2004). Preeclampsia and future cardiovascular disease: potential role of altered angiogenesis and insulin resistance. The Journal of Clinical Endocrinology and Metabolism, 89, 6239–6243.
Bello, N., Rendon, I. S. H., & Arany, Z. (2013). The relationship between pre-eclampsia and peripartum cardiomyopathy: a systematic review and meta-analysis. Journal of the American College of Cardiology, 62(18), 1715–1723.
Ntusi, N. B. A., Badri, M., Gumedze, F., Sliwa, K., & Mayosi, B. M. (2015). Pregnancy-associated heart failure: a comparison of clinical presentation and outcome between hypertensive heart failure of pregnancy and idiopathic peripartum cardiomyopathy. PloS One, 10(8), e0133466. doi:10.1371/journal.pone.0133466.
Billieux, M.-H., Petignat, P., Fior, A., Mhawech, P., Blouin, J.-L., Dahoun, S., & Vassilakos, P. (2004). Pre-eclampsia and peripartum cardiomyopathy in molar pregnancy: clinical implication for maternally imprinted genes. Ultrasound in Obstetrics & Gynecology, 23, 398–401.
Gleicher, N., & Elkayam, U. (2009). Peripartum cardiomyopathy, an autoimmune manifestation of allograft rejection? Autoimmunity Reviews, 8, 384–387.
Ponikowski, P., Voors, A. A., Anker, S. D., Bueno, H., Cleland, J. G., Coats, A. J., et al. (2016). 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). European Heart Journal, 37(27), 2129–2200.
Yancy, C. W., Jessup, M., Bozkurt, B., Butler, J., Casey Jr., D. E., Drazner, M. H., et al. (2013). 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Journal of the American College of Cardiology, 62(16), e147–e239.
Hershberger, R. E., Lindenfeld, J., Mestroni, L., Seidman, C. E., Taylor, M. R., & Towbin, J. A. (2009). Genetic evaluation of cardiomyopathy—a Heart Failure Society of America practice guideline. Journal of Cardiac Failure, 15(2), 83–97.
Wilson, K. D., Shen, P., Fung, E., Karakikes, I., Zhang, A., InanlooRahatloo, K., et al. (2015). A rapid, high-quality, cost-effective, comprehensive and expandable targeted next-generation sequencing assay for inherited heart diseases. Circulation Research, 117(7), 603–611.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Dr. Lee has no potential conflicts of interest. Dr. Judge has received payment as a scientific advisor to Alnylam, Array Biopharma, GlaxoSmithKline, Invitae, MyoKardia, and Pfizer. This review article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Associate Editor Paul J. R. Barton oversaw the review of this article
Rights and permissions
About this article

Cite this article
Lee, Y.Z.J., Judge, D.P. The Role of Genetics in Peripartum Cardiomyopathy. J. of Cardiovasc. Trans. Res. 10, 437–445 (2017). https://doi.org/10.1007/s12265-017-9764-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12265-017-9764-y