
Abstract
G protein coupled receptor kinase type 2 (GRK2) plays an important role in the development and maintenance of cardiac hypertrophy and heart failure even if its exact role is still unknown. In this study, we assessed the effect of GRK2 on the regulation of cardiac hypertrophy. In H9C2 cells, GRK2 overexpression increased atrial natriuretic factor (ANF) activity and enhanced phenylephrine-induced ANF response, and this is associated with an increase of NFκB transcriptional activity. The kinase dead mutant and a synthetic inhibitor of GRK2 activity exerted the opposite effect, suggesting that GRK2 regulates hypertrophy through upregulation of NFκB activity in a phosphorylation-dependent manner. In two different in vivo models of left ventricle hypertrophy (LVH), the selective inhibition of GRK2 activity prevented hypertrophy and reduced NFκB transcription activity. Our results suggest a previously undisclosed role for GRK2 in the regulation of hypertrophic responses and propose GRK2 as potential therapeutic target for limiting LVH.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
G protein coupled receptor (GPCR) kinases (GRKs) are serine/threonine kinases which regulate the activity of GPCR through phosphorylation [1]. Changes in GRK activity and expression have been shown to play an important role in the development and maintenance of cardiac hypertrophy and heart failure [2–4] even if their exact role remains to be elucidated. In particular, GRK2 is known to be regulated in several cardiovascular disorders since increased levels of GRK2 are observed during left ventricular hypertrophy (LVH) [3, 4, 5, 6, 7] and are associated with an impaired cardiac contractility via reduction of beta adrenergic receptor inotropic response [2].
As to the mechanistic role of GRK2 in the heart, it has been shown that, besides βAR downregulation and desensitization, GRK2 can engage with different intracellular partners to regulate several cardiomyocyte functions. Indeed, this kinase induces cardiac insulin resistance and reduces cardiac metabolic plasticity [8], regulates mitochondrial function [9, 10], and participates to intracellular calcium homeostasis [11]. Given its molecular and functional complexity, it is not surprising that this molecule has a pivotal role in regulating both function and development of cardiovascular system. Indeed, global genetic deletion of GRK2 is lethal by preventing the correct development of cardiovascular system in prenatal life [12] and in adult life the selective cardiac deletion of GRK2 leads to a prevalent eccentric remodeling after chronic exposure to β adrenergic stimulation [13]. This evidence suggests that GRK2 might participate to the hypertrophic responses of the injured adult heart.
The molecular mechanisms underlying LVH comprise the activation of several transcription factors, such as MEF-2, GATA-4, and NFAT [14–18]. Recently, nuclear factor-κB (NFκB) has also been associated with the development of cardiac hypertrophy [19, 20]. Indeed, NFκB has been demonstrated to be relevant in the pathophysiology of LVH and remodeling through mechanisms independent from inflammation [21]. NFκB mediates hypertrophic growth of cardiomyocytes in response to G protein coupled receptor (GPCR) agonists, including norepinephrine, endothelin-1, and angiotensin-II [22, 23]. Additionally, NFκB inhibition has been shown to attenuate LVH in different animal models [24, 25]. This evidence suggests that NFκB blockade may be an effective strategy to counteract LVH and cardiac remodeling [25, 26].
Based on such a background, the aim of this study is to evaluate the role of GRK2 in the development of LVH both in vitro and in vivo and whether such phenomenon is associated with transcription factors relevant to LVH, in particular NFκB activity.
Methods
Cell Culture
A cell line of cardiac myoblasts (H9C2), which is a well-established model of hypertrophy in vitro [27–30], was maintained in culture in DMEM supplemented with 10 % FBS at 37 °C in 95 % air–5 % CO2.
Luciferase Assay
Cells were transfected with plasmid expression vectors containing the luciferase reporter gene linked to five repeats of an NFκB binding site (κB-Luc) or atrial natriuretic factor (ANF) promoter. Lysates were analyzed using the luciferase assay system with reporter lysis buffer from Promega.
Immunoprecipitation and Western Blot
Cells were lysed in RIPA/sodium dodecyl sulfate (SDS) buffer. Endogenous IκBα or GRK2 from total extracts were immunoprecipitated with specific antibodies (Santa Cruz) and protein A/G agarose (Santa Cruz). After extensive washing, the immunocomplexes were electrophoresed by SDS/PAGE and transferred to nitrocellulose. For western blot analysis, the experiments were performed as described previously [25, 31, 32].
Electrophoretic Mobility Shift Assay
Nuclear proteins were isolated from heart samples, and NFκB binding activity was examined by electrophoretic mobility shift assay, as described previously [31, 33].
In Vivo Study
Experiments were carried out on 12-week-old normotensive Wistar Kyoto (WKY) and spontaneously hypertensive male rats (Charles River, Calco, LC, Italy). Four injections (50 μL each) of Ant-124 (10−4 M) or Ant as control, into the cardiac wall, were performed once a week for 3 weeks. In the WKY group, after cardiac injection, we implanted subcutaneously a miniosmotic pump (ALZET 2004) releasing phenylephrine (PE) (100 mg/kg).
Echocardiography
Transthoracic echocardiography was performed at days 0, 7, 14, and 21 after surgery using a dedicated small-animal high-resolution imaging system (VeVo 770, Visualsonics, Inc).
Isolation of Ventricular Cardiomyocytes and Real-Time PCR
Adult mouse ventricular myocytes were isolated from GRK2fl/fl mice hearts by a standard enzymatic digestion procedure as previously described [34]. Total RNA was isolated and real-time quantitative PCR was performed.
Statistical Analysis
All data are presented as mean ± SEM. Two-way ANOVA with a Bonferroni post hoc test was performed to compare the different parameters between the different groups. A p value <0.05 was considered significant. Statistical analysis was performed using GraphPad Prism version 5.01 (GraphPad Software, Inc., San Diego, CA).
Results
GRK2 Induces Hypertrophy In Vitro
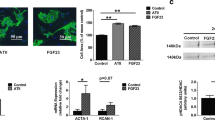
To evaluate the ability of GRK2 to modulate hypertrophy in vitro, we performed an ANF-promoter-driven luciferase assay to evaluate ANF promoter activity and real-time PCR to evaluate ANF gene expression. In H9C2, the overexpression of GRK2 was performed by transient transfection, and hypertrophy was induced by stimulation with phenylephrine (PE). GRK2 overexpression increased ANF promoter activity (Fig. 1a) and ANF gene expression (Fig. 1b) and enhanced the response to PE. To confirm this finding, we also evaluated BNP gene expression, another marker of hypertrophy, by real-time PCR. GRK2 overexpression increased BNP gene expression and enhanced the response to PE (Supplementary Fig. S1). To evaluate whether the catalytic activity of GRK2 is needed to induce such phenomenon, we overexpressed in cells the kinase dead mutant of GRK2 (GRK2-DN). Overexpression of GRK2-DN inhibited ANF promoter activity (Fig. 1a) and ANF gene expression (Fig. 1b) both basally and in response to PE, opposite to the naive kinase. Similar results were found in the regulation of BNP gene expression (Supplementary Fig. S1). To further support these data, we treated cells with a previously described peptide inhibitor of GRK2, Ant-124 [35]. As expected, Ant-124 inhibits the catalytic activity of GRK2 in cardiomyoblasts (Fig. 1c). Interestingly, the treatment with Ant-124 inhibited both PE- and GRK2-induced ANF promoter activity (Fig. 1d) and ANF gene expression (Fig. 1e) compared with Ant. To evaluate whether GRK2 inhibition was effective also in response to other hypertrophic stimuli, besides alpha-adrenergic activation, we evaluated ANF gene expression in response to angiotensin II. The inhibition of GRK2 was effective also in response to angiotensin II (Supplementary Fig. S2).

GRK2 regulates hypertrophy in vitro (a–e). To assess the role of GRK2 in hypertrophy, we evaluated ANF promoter activity by luciferase assay and ANF gene expression by real-time PCR. H9C2 cells were transfected with an empty plasmid (CTRL) and a plasmid encoding GRK2 or GRK2-DN. The overexpression of GRK2 induces ANF promoter activity (a) and ANF gene expression (b) both basally and in response to PE. The overexpression of a kinase dead mutant of GRK2 inhibits ANF promoter activity in response to PE with respect to the wild type form of GRK2. To confirm such finding, we use a synthetic inhibitor of GRK2, Ant-124, that specifically inhibits its kinase activity (c). Ant-124 significantly inhibits ANF promoter activity (d) and ANF gene expression (e) in response to PE and following GRK2 overexpression (d). The scramble peptide Ant was used as control. Results are the mean of five independent experiments. *p < 0.05 versus untreated CTRL; # p < 0.05 versus PE-CTRL; § p < 0.05 versus GRK2
GRK2 Induces Hypertrophy by Regulating NFκB Activity
In other cell types, GRK2 deletion or overexpression reciprocally regulates TNFα-induced IκBα phosphorylation and degradation and consequent regulation of NFκB [36]. Thus, we evaluated the possibility that GRK2 could regulate the activation of NFκB signaling also in H9C2. To this aim, we first confirmed that GRK2 regulates IκBα levels. Indeed, overexpression of GRK2 reduced IκBα levels while GRK2-DN has no effects (Fig. 2a). We then evaluated if GRK2 was able to interact with and phosphorylate IκBα as previously demonstrated for GRK5 [31]. Figure 2b shows that GRK2 co-precipitates with IκBα in whole cell lysates, and results of the kinase assay with purified proteins shows that GRK2 is also able to phosphorylate IκBα (Fig. 2c). Consistently, the overexpression of GRK2-WT increased NFκB activity both basally and in response to PE; GRK2-DN and Ant-124 reduced such response (Fig. 2d). To confirm that GRK2 regulates hypertrophic responses by inducing NFκB activity, we evaluated the effects of an inhibitor of NFκB, IκBα, on GRK2-dependent ANF promoter activity in response to PE. The overexpression of IκBα inhibited PE-induced ANF promoter activity both basally and in response to GRK2 overexpression (Fig. 2e). Altogether, these data suggest that GRK2 can regulate hypertrophic responses in vitro by modulating NFκB activity through the phosphorylation of IκBα.

GRK2 induces hypertrophy by regulating NFκB activity. a In whole cell lysates from H9C2, IκBα levels were analyzed by immunoblot. The overexpression of GRK2 reduces total levels of IκBα while the kinase dead mutant does not modify IκBα levels with respect to control. Actin was used as loading control and GRK2 levels were evaluated to verify whether transfection occurred. Images are representative of three independent experiments. b The ability of GRK2 to precipitate IκBα was evaluated by co-immunoprecipitation and western blot. GRK2 is able to precipitate IκBα and the overexpression of the kinase enhances such binding while GRK2DN has no effect. Images are representative of three independent experiments. c To assess the ability of GRK2 to phosphorylate IκBα, we performed a kinase activity assay. GRK2 is able to phosphorylate itself and to phosphorylate IκBα. IκBα alone was used as negative control. Images are the result of three independent experiments. d To evaluate whether GRK2 could regulate NFκB activity, we performed a luciferase assay. GRK2 is able to induce NFκB activity both basally and in response to PE while both GRK2-DN and Ant-124 prevent such activation. The scramble peptide Ant was used as control. Results are the mean of five independent experiments. *p < 0.05 versus CTRL; # p < 0.05 versus PE. e We evaluated the effects of an inhibitor of NFκB, IκBα, on GRK2-dependent ANF promoter activity in response to PE. The overexpression of IκBα inhibited PE-induced ANF promoter activity both basally and in response to GRK2 overexpression. Results are the mean of five independent experiments. *p < 0.05 versus CTRL; # p < 0.05 versus PE
The Knockdown of GRK2 Reduces Hypertrophic Responses by Inhibiting NFκB Activity
In order to confirm the involvement of GRK2 in the regulation of hypertrophic phenotypes in vitro, we evaluated the effects of GRK2 knockdown on ANF gene expression in response to PE. In H9C2 cells, the knockdown of GRK2 was induced by means of a specific siRNA, as previously described [10], leading to a significant reduction of GRK2 expression with respect to the transfection of a scramble siRNA (Fig. 3a). GRK2 knockdown is associated with a significant increase of IκB levels (Fig. 3a) and a reduction of ANF gene expression in response to PE (Fig. 3b). These data confirmed the key role of GRK2 in the development of PE-induced hypertrophic phenotype in vitro.

The knockdown of GRK2 reduces hypertrophic responses by inhibiting NFκB activity. a, b In H9C2 cells, the knockdown of GRK2 was induced by means of a specific siRNA. Control and PE were treated with a scramble siRNA. GRK2 and IκB levels were evaluated by immunoblot. GRK2 downregulation, confirmed by the reduction of GRK2 expression respect to the scramble transfection (CTRL), associates with an increase of IκB levels (a). GRK2 knockdown causes a reduction of ANF gene expression in response to PE, evaluated by real-time PCR (b). *p < 0.05 versus CTRL; # p < 0.05 versus PE
GRK2 Regulates the Hypertrophic Phenotype in SHR Rats
To confirm the physiological relevance of the in vitro results, we evaluated the role of GRK2 in the development of cardiac hypertrophy in spontaneously hypertensive (SHR) rats, an animal model of hypertension-induced LVH. To this aim, the inhibitor of GRK2, Ant-124, or the scramble peptide, Ant, was injected in the cardiac wall of SHR, and echocardiographic analysis was performed weekly to evaluate the progression of hypertrophy (Supplementary Fig. S3). At the end of the treatment (21 days), left ventricular mass (LVM) was enhanced in SHR with respect to WKY and the treatment with Ant-124 significantly reduced such values (Fig. 4a). A physiological reduction of the left ventricular end diastolic diameter (LVEDD) was observed in SHR compared with WKY, which was recovered in SHR treated with Ant-124 (Fig. 4b). Intraventricular septal (IVS) was thicker in SHR and the treatment with Ant-124 reduced such value (Fig. 4c). This is associated with an ameliorated cardiac function in SHR treated with Ant-124 compared with controls (Fig. 4d). The analysis of echocardiographic parameters show that SHR is a model of concentric hypertrophy, according the Knoll et al. paper [37], and that the inhibition of the catalytic activity of GRK2 in this model protects from left ventricular hypertrophy suggesting that this kinase is deleterious in the development of such pathological condition. Indeed, GRK2 levels in hearts from SHR are significantly higher with respect to WKY and were partially reduced by the treatment with Ant-124 (Fig. 4e). To confirm that GRK2-dependent LVH is strictly associated with the ability of the kinase to regulate NFκB activity, we evaluated NFκB activity by EMSA in hearts from control and treated SHR. Figure 4f shows that NFκB activity was enhanced in SHR hearts with respect to WKY and the treatment with Ant-124 significantly reduced such activation.

The inhibition of GRK2 by Ant-124 activity reduces hypertrophy in SHR. a–d SHR rats were treated with Ant-124 or the scramble peptide Ant by intramyocardial injection, and echocardiographic parameters were evaluated to analyze the effect of Ant-124 on the progression of cardiac hypertrophy. At the end of the treatment, LVM (a) was significantly reduced in rats treated with Ant-124 compared with those treated with Ant. A reduction of LVEDD was observed in SHR compared with WKY, which was recovered in SHR treated with Ant-124 (b). IVS (c) was enhanced in SHR rats and reduced by Ant-124. Accordingly, cardiac function was enhanced in treated rats (d). *p < 0.05 versus WKY; # p < 0.05 versus SHR vehicle. e GRK2 levels were evaluated by immunoblot in hearts from the different groups. GRK2 levels increased in SHR with respect to WKY, and the treatment with Ant-124 reduced such effect. f Nuclear extracts were prepared from collected hearts, and NFκB activity was evaluated by EMSA. The ability of NFκB to bind DNA was significantly increased in SHR with respect to WKY, and Ant-124 treatment prevented such an increase. Densitometric analysis is shown in graph. *p < 0.05 versus WKY; # p < 0.05 versus SHR Vehicle. NC negative control
GRK2 Is Involved in the Development of the Hypertrophic Phenotype in WKY Rats
The SHR model of hypertrophy suggests that GRK2 is involved in the progression of cardiac hypertrophy. To assess whether GRK2 is involved in the development of LVH, WKY rats were treated with intramyocardial injection of Ant-124 or Ant before inducing hypertrophy by chronic administration of PE (14 days) using mini-osmotic pumps. Echocardiographic analysis was performed at the end of the treatment to evaluate the progression of hypertrophy (Supplementary Fig. S4). According the Knoll et al. paper [37], WKY rats treated with chronic infusion of PE present a concentric hypertrophy since there is an increase of cardiac size (Fig. 5a) and IVS (Fig. 5b) and a reduction of LVEDD (Fig. 5c). Ant-124 prevented the progression of cardiac hypertrophy since both LVM (Fig. 5a) and IVS (Fig. 5b) were reduced in PE-treated rats with respect to controls and LVEDD was recovered (Fig. 5c). These data suggest that the inhibition of GRK2 activity prevents PE-induced cardiac hypertrophy. To confirm this result, we evaluated IκBα levels in hearts collected from treated rats. Figure 5c shows that the inhibition of GRK2 activity increased IκBα levels both basally and in response to PE. GRK2 levels were increased in response to PE and reduced by the treatment with Ant-124 (Fig. 5c).

Ant-124 inhibits the development of PE-induced hypertrophy. a–d WKY rats were treated with Ant-124 or Ant by intramyocardial injection, and then hypertrophy was induced through infusion of PE by osmotic pumps. Echocardiographic parameters were evaluated to analyze the effect of Ant-124 on the development of cardiac hypertrophy. Both LVM (a) and IVS (b) were significantly reduced in rats treated with Ant-124 compared with those treated with Ant. A reduction of LVEDD (c) was observed in WKY treated with PE compared with controls that was recovered by the treatment with Ant-124. *p < 0.05 versus untreated; # p < 0.05 versus PE vehicle. d IκBα and GRK2 levels were evaluated in hearts collected from treated rats. IκBα levels were increased in treated hearts in response to PE. GRK2 levels were increased in response to PE and reduced by the treatment. Actin was used as loading control. Results are the mean of five independent experiments
The Knockdown of GRK2 Inhibits Cardiac Hypertrophy In Vivo
Hypertrophy was induced in GRK2fl/fl mice through means of implantation of miniosmotic pumps which gradually release PE. A group of mice was treated with an intracardiac injection of an adenovirus coding for CRE recombinase (the enzyme that catalyzes site-specific recombination of DNA between two loxP sites) to induce GRK2 knockout in the heart, confirmed by immunoblot (Fig. 6a). Echocardiographic parameters were evaluated after 2 weeks from starting treatment. In particular, IVS (Fig. 6b) and LVM (Fig. 6c) were reduced in CRE-treated GRK2fl/fl mice with respect to control GRK2fl/fl mice in response to PE. Accordingly, PE-induced impairment of cardiac function was restored by GRK2 knockdown (Fig. 6d), and this is associated with a reduction of ANF gene expression in cardiac myocytes (Fig. 6e).

The knockdown of GRK2 inhibits cardiac hypertrophy in vivo. a GRK2fl/fl mice were treated with PE, and an intramyocardial injection of an adenovirus coding for CRE or an empty adenovirus as control was performed to induce GRK2 knockout in the heart, as assessed by immunoblot analysis. b–d Echocardiographic parameters were evaluated after 2 weeks from starting treatment. IVS (b) and LVM (c) were reduced in CRE-treated GRK2fl/fl mice with respect to control GRK2fl/fl mice in response to PE. Accordingly, PE-induced impairment of cardiac function was restored by GRK2 silencing (d). *p < 0.05 versus CTRL; # p < 0.05 versus PE Vehicle. e Cardiac myocytes were collected from hearts of GRK2fl/fl mice treated with CRE and PE. ANF gene expression was evaluated by real-time PCR. The cardiac knockout of GRK2 in GRK2fl/fl mice treated with CRE reduced ANF gene expression in response to PE compared with controls. *p < 0.05 versus CTRL; # p < 0.05 versus PE vehicle
Discussion
Our study produces three steps forward in the knowledge of the molecular basis of LVH leading to the identification of new targets for therapeutic treatments. First of all, we identify a new cytosolic substrate of GRK2 in the heart, IκBα. Second, we demonstrate that GRK2-dependent regulation of NFκB transcription activity contributes to the development of LVH. Third, our study proposes a new therapeutic strategy for the treatment of LVH based on the downregulation of the expression levels of GRK2 or the specific inhibition of its catalytic activity by a new small molecule, Ant-124.
Our data identify a new non-canonical role of GRK2 in the heart which is based on the phosphorylation of a novel cytosolic partner, IκBα, leading to the regulation of hypertrophy-related gene expression. Thus, the present study defines a new role of GRK2 in cardiac hypertrophy and identifies NFκB as the intrinsic mechanism. The ability to regulate transcription factors in hypertrophy has been recently demonstrated for GRK5 [38] which, by acting in a kinase-independent manner, regulates NFAT activity and LVH [38]. Here we demonstrate for the first time that also GRK2 participates to the setting of the hypertrophic phenotype through the regulation of NFκB. This latter is not a specific cardiac transcription factor, but its involvement in the regulation of LVH is extensively demonstrated [25]. Indeed, we demonstrated that NFκB activity is enhanced in LVH and its inhibition prevents the development of cardiac hypertrophy and ameliorates cardiac function [25]. Here we show that GRK2 is able to bind and phosphorylate IκBα, the NFκB inhibitor, leading to the activation of NFκB transcriptional activity and consequently to the increase of the hypertrophic gene expression, ANF. This effect is abolished by GRK2 knockdown both in vitro and in vivo. The activity of the kinase is needed to regulate hypertrophic phenotypes. Indeed, in different animal models of hypertrophy (hypertension-induced hypertrophy in SHR and PE-induced hypertrophy in WKY), pharmacological inhibition of GRK2 catalytic activity through a synthetic inhibitor, Ant-124, reduces LVH. It has already been described that the inhibition of GRK2 activity is advantageous for the treatment of cardiovascular diseases, such as heart failure [39], but it has never been shown in LVH and never with a pharmacological inhibitor. Until now, the inhibition of GRK2 activity was induced by means of a GRK2 truncated mutant containing the carboxy-terminal domain of the kinase (GRK2-ct), which really acts as a scavenger of GRK2 from plasma membrane rather than an inhibitor of the kinase activity. Here, we used a small peptide that is more selective than GRK2-ct since it specifically binds the HJ loop of GRK2 catalytic domain leading to the selective inhibition of the kinase activity [35] without affecting other pathways which are independent from GRK2. This latter aspect is advantageous for a therapeutic treatment since the effects of GRK2-ct depends on its interaction with Gβγ which causes not only the inhibition of GRK2 signaling on plasma membrane but also the inhibition of specific Gβγ signaling. GRK2-ct bears therefore the ability to interact with other several signal transduction pathways within the cell, such as MAPK and PI3K signaling, increasing the possibility of undesired effects.
In conclusion, our results suggest a novel role of GRK2 in the regulation of hypertrophy both in vitro and in vivo that is in accord with recent literature which shows that GRK2 downregulation/inhibition is useful to ameliorate cardiac function in pathological conditions, such as insulin resistance, diabetes, or obesity [40]. Moreover, here we propose the downregulation of GRK2 levels or the inhibition of its catalytic activity as a new strategy to ameliorate cardiac function in hypertrophy. In particular, we propose Ant-124 as a prototype for the generation of a pharmacophore to use for therapeutic applications for the treatment of LVH.
References
Ferguson, S. S. (2001). Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacological Reviews, 53(1), 1–24 [Research Support, Non-U.S. Gov’t Review].
Choi, D. J., Koch, W. J., Hunter, J. J., & Rockman, H. A. (1997). Mechanism of beta-adrenergic receptor desensitization in cardiac hypertrophy is increased beta-adrenergic receptor kinase. Journal of Biological Chemistry, 272(27), 17223–17229 [Research Support, U.S. Gov’t, P.H.S.].
Anderson, K. M., Eckhart, A. D., Willette, R. N., & Koch, W. J. (1999). The myocardial beta-adrenergic system in spontaneously hypertensive heart failure (SHHF) rats. Hypertension, 33(1 Pt 2), 402–407.
Ungerer, M., Bohm, M., Elce, J. S., Erdmann, E., & Lohse, M. J. (1993). Altered expression of beta-adrenergic receptor kinase and beta 1-adrenergic receptors in the failing human heart. Circulation, 87(2), 454–463 [Research Support, Non-U.S. Gov’t].
Ungerer, M., Parruti, G., Bohm, M., Puzicha, M., DeBlasi, A., Erdmann, E., et al. (1994). Expression of beta-arrestins and beta-adrenergic receptor kinases in the failing human heart. Circulation Research, 74(2), 206–213 [Research Support, Non-U.S. Gov’t].
Ping, P., Anzai, T., Gao, M., & Hammond, H. K. (1997). Adenylyl cyclase and G protein receptor kinase expression during development of heart failure. American Journal of Physiology, 273(2 Pt 2), H707–717 [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, Non-P.H.S. Research Support, U.S. Gov’t, P.H.S.].
Iaccarino, G., Barbato, E., Cipolletta, E., De Amicis, V., Margulies, K. B., Leosco, D., et al. (2005). Elevated myocardial and lymphocyte GRK2 expression and activity in human heart failure. European Heart Journal, 26(17), 1752–1758. doi:10.1093/eurheartj/ehi429 [Multicenter Study Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.].
Woodall, M. C., Ciccarelli, M., Woodall, B. P., & Koch, W. J. (2014). G protein-coupled receptor kinase 2: a link between myocardial contractile function and cardiac metabolism. Circulation Research, 114(10), 1661–1670. doi:10.1161/CIRCRESAHA.114.300513 [Research Support, N.I.H., Extramural Review].
Fusco, A., Santulli, G., Sorriento, D., Cipolletta, E., Garbi, C., Dorn, G. W., 2nd, et al. (2012). Mitochondrial localization unveils a novel role for GRK2 in organelle biogenesis. Cellular Signalling, 24(2), 468–475. doi:10.1016/j.cellsig.2011.09.026 [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t].
Sorriento, D., Fusco, A., Ciccarelli, M., Rungi, A., Anastasio, A., Carillo, A., et al. (2013). Mitochondrial G protein coupled receptor kinase 2 regulates proinflammatory responses in macrophages. FEBS Letters, 587(21), 3487–3494. doi:10.1016/j.febslet.2013.09.002 [Research Support, Non-U.S. Gov’t].
Raake, P. W., Zhang, X., Vinge, L. E., Brinks, H., Gao, E., Jaleel, N., et al. (2012). Cardiac G-protein-coupled receptor kinase 2 ablation induces a novel Ca2+ handling phenotype resistant to adverse alterations and remodeling after myocardial infarction. Circulation, 125(17), 2108–2118. doi:10.1161/CIRCULATIONAHA.111.044255 [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t].
Raake, P. W., Vinge, L. E., Gao, E., Boucher, M., Rengo, G., Chen, X., et al. (2008). G protein-coupled receptor kinase 2 ablation in cardiac myocytes before or after myocardial infarction prevents heart failure. Circulation Research, 103(4), 413–422. doi:10.1161/CIRCRESAHA.107.168336 [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t].
Matkovich, S. J., Diwan, A., Klanke, J. L., Hammer, D. J., Marreez, Y., Odley, A. M., et al. (2006). Cardiac-specific ablation of G-protein receptor kinase 2 redefines its roles in heart development and beta-adrenergic signaling. Circulation Research, 99(9), 996–1003. doi:10.1161/01.RES.0000247932.71270.2c [Research Support, N.I.H., Extramural].
Molkentin, J. D. (2000). Calcineurin and beyond: cardiac hypertrophic signaling. Circulation Research, 87(9), 731–738 [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. Review].
Kuo, C. T., Morrisey, E. E., Anandappa, R., Sigrist, K., Lu, M. M., Parmacek, M. S., et al. (1997). GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes and Development, 11(8), 1048–1060 [Research Support, U.S. Gov’t, P.H.S.].
Molkentin, J. D. (2004). Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovascular Research, 63(3), 467–475. doi:10.1016/j.cardiores.2004.01.021 [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. Review].
Molkentin, J. D., & Markham, B. E. (1993). Myocyte-specific enhancer-binding factor (MEF-2) regulates alpha-cardiac myosin heavy chain gene expression in vitro and in vivo. Journal of Biological Chemistry, 268(26), 19512–19520 [Comparative Study Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.].
Pikkarainen, S., Tokola, H., Majalahti-Palviainen, T., Kerkela, R., Hautala, N., Bhalla, S. S., et al. (2003). GATA-4 is a nuclear mediator of mechanical stretch-activated hypertrophic program. Journal of Biological Chemistry, 278(26), 23807–23816. doi:10.1074/jbc.M302719200 [Research Support, Non-U.S. Gov’t].
Purcell, N. H., Tang, G., Yu, C., Mercurio, F., DiDonato, J. A., & Lin, A. (2001). Activation of NF-kappa B is required for hypertrophic growth of primary rat neonatal ventricular cardiomyocytes. Proceedings of the National Academy of Sciences of the United States of America, 98(12), 6668–6673. doi:10.1073/pnas.111155798 [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.].
Hirotani, S., Otsu, K., Nishida, K., Higuchi, Y., Morita, T., Nakayama, H., et al. (2002). Involvement of nuclear factor-kappaB and apoptosis signal-regulating kinase 1 in G-protein-coupled receptor agonist-induced cardiomyocyte hypertrophy. Circulation, 105(4), 509–515 [Research Support, Non-U.S. Gov’t].
Kawamura, N., Kubota, T., Kawano, S., Monden, Y., Feldman, A. M., Tsutsui, H., et al. (2005). Blockade of NF-kappaB improves cardiac function and survival without affecting inflammation in TNF-alpha-induced cardiomyopathy. Cardiovascular Research, 66(3), 520–529. doi:10.1016/j.cardiores.2005.02.007 [Research Support, Non-U.S. Gov’t].
Aikawa, R., Nagai, T., Tanaka, M., Zou, Y., Ishihara, T., Takano, H., et al. (2001). Reactive oxygen species in mechanical stress-induced cardiac hypertrophy. Biochemical and Biophysical Research Communications, 289(4), 901–907. doi:10.1006/bbrc.2001.6068 [Research Support, Non-U.S. Gov’t].
Harada, K., Komuro, I., Shiojima, I., Hayashi, D., Kudoh, S., Mizuno, T., et al. (1998). Pressure overload induces cardiac hypertrophy in angiotensin II type 1A receptor knockout mice. Circulation, 97(19), 1952–1959 [Research Support, Non-U.S. Gov’t].
Li, Y., Ha, T., Gao, X., Kelley, J., Williams, D. L., Browder, I. W., et al. (2004). NF-kappaB activation is required for the development of cardiac hypertrophy in vivo. American Journal of Physiology - Heart and Circulatory Physiology, 287(4), H1712–1720. doi:10.1152/ajpheart.00124.2004 [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.].
Sorriento, D., Santulli, G., Fusco, A., Anastasio, A., Trimarco, B., & Iaccarino, G. (2010). Intracardiac injection of AdGRK5-NT reduces left ventricular hypertrophy by inhibiting NF-kappaB-dependent hypertrophic gene expression. Hypertension, 56(4), 696–704. doi:10.1161/HYPERTENSIONAHA.110.155960 [Research Support, Non-U.S. Gov’t].
Kawano, S., Kubota, T., Monden, Y., Kawamura, N., Tsutsui, H., Takeshita, A., et al. (2005). Blockade of NF-kappaB ameliorates myocardial hypertrophy in response to chronic infusion of angiotensin II. Cardiovascular Research, 67(4), 689–698. doi:10.1016/j.cardiores.2005.04.030 [Research Support, Non-U.S. Gov’t].
Jeong, S., & Yoon, M. (2009). Fenofibrate inhibits adipocyte hypertrophy and insulin resistance by activating adipose PPARalpha in high fat diet-induced obese mice. Experimental and Molecular Medicine, 41(6), 397–405. doi:10.3858/emm.2009.41.6.045 [Research Support, Non-U.S. Gov’t].
Watkins, S. J., Borthwick, G. M., & Arthur, H. M. (2011). The H9C2 cell line and primary neonatal cardiomyocyte cells show similar hypertrophic responses in vitro. In Vitro Cellular and Developmental Biology - Animal, 47(2), 125–131. doi:10.1007/s11626-010-9368-1 [Research Support, Non-U.S. Gov’t].
Verma, S. K., Krishnamurthy, P., Barefield, D., Singh, N., Gupta, R., Lambers, E., et al. (2012). Interleukin-10 treatment attenuates pressure overload-induced hypertrophic remodeling and improves heart function via signal transducers and activators of transcription 3-dependent inhibition of nuclear factor-kappaB. Circulation, 126(4), 418–429. doi:10.1161/CIRCULATIONAHA.112.112185 [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t].
Chen, Q. M., Tu, V. C., Wu, Y., & Bahl, J. J. (2000). Hydrogen peroxide dose dependent induction of cell death or hypertrophy in cardiomyocytes. Archives of Biochemistry and Biophysics, 373(1), 242–248. doi:10.1006/abbi.1999.1558 [Research Support, Non-U.S. Gov’t].
Sorriento, D., Ciccarelli, M., Santulli, G., Campanile, A., Altobelli, G. G., Cimini, V., et al. (2008). The G-protein-coupled receptor kinase 5 inhibits NFkappaB transcriptional activity by inducing nuclear accumulation of IkappaB alpha. Proceedings of the National Academy of Sciences of the United States of America, 105(46), 17818–17823. doi:10.1073/pnas.0804446105.
Iaccarino, G., Izzo, R., Trimarco, V., Cipolletta, E., Lanni, F., Sorriento, D., et al. (2006). Beta2-adrenergic receptor polymorphisms and treatment-induced regression of left ventricular hypertrophy in hypertension. Clinical Pharmacology and Therapeutics, 80(6), 633–645. doi:10.1016/j.clpt.2006.09.006 [Research Support, Non-U.S. Gov’t].
Sorriento, D., Campanile, A., Santulli, G., Leggiero, E., Pastore, L., Trimarco, B., et al. (2009). A new synthetic protein, TAT-RH, inhibits tumor growth through the regulation of NFkappaB activity. Molecular Cancer, 8, 97. doi:10.1186/1476-4598-8-97.
Ciccarelli, M., Chuprun, J. K., Rengo, G., Gao, E., Wei, Z., Peroutka, R. J., et al. (2011). G protein-coupled receptor kinase 2 activity impairs cardiac glucose uptake and promotes insulin resistance after myocardial ischemia. Circulation, 123(18), 1953–1962. doi:10.1161/CIRCULATIONAHA.110.988642 [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t].
Cipolletta, E., Campanile, A., Santulli, G., Sanzari, E., Leosco, D., Campiglia, P., et al. (2009). The G protein coupled receptor kinase 2 plays an essential role in beta-adrenergic receptor-induced insulin resistance. Cardiovascular Research, 84(3), 407–415. doi:10.1093/cvr/cvp252 [Research Support, Non-U.S. Gov’t].
Patial, S., Luo, J., Porter, K. J., Benovic, J. L., & Parameswaran, N. (2010). G-protein-coupled-receptor kinases mediate TNFalpha-induced NFkappaB signalling via direct interaction with and phosphorylation of IkappaBalpha. Biochemical Journal, 425(1), 169–178. doi:10.1042/BJ20090908 [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t].
Knoll, R., Iaccarino, G., Tarone, G., Hilfiker-Kleiner, D., Bauersachs, J., Leite-Moreira, A. F., et al. (2011). Towards a re-definition of ‘cardiac hypertrophy’ through a rational characterization of left ventricular phenotypes: a position paper of the Working Group ‘Myocardial Function’ of the ESC. European Journal of Heart Failure, 13(8), 811–819. doi:10.1093/eurjhf/hfr071 [Practice Guideline Research Support, Non-U.S. Gov’t].
Hullmann, J. E., Grisanti, L. A., Makarewich, C. A., Gao, E., Gold, J. I., Chuprun, J. K., et al. (2014). GRK5-Mediated Exacerbation of Pathological Cardiac Hypertrophy Involves Facilitation of Nuclear NFAT Activity. Circulation Research, 115(12), 976–985. doi:10.1161/CIRCRESAHA.116.304475.
Raake, P. W., Schlegel, P., Ksienzyk, J., Reinkober, J., Barthelmes, J., Schinkel, S., et al. (2013). AAV6.betaARKct cardiac gene therapy ameliorates cardiac function and normalizes the catecholaminergic axis in a clinically relevant large animal heart failure model. European Heart Journal, 34(19), 1437–1447. doi:10.1093/eurheartj/ehr447 [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t].
Lucas, E., Jurado-Pueyo, M., Fortuno, M. A., Fernandez-Veledo, S., Vila-Bedmar, R., Jimenez-Borreguero, L. J., et al. (2014). Downregulation of G protein-coupled receptor kinase 2 levels enhances cardiac insulin sensitivity and switches on cardioprotective gene expression patterns. Biochimica et Biophysica Acta, 1842(12 Pt A), 2448–2456. doi:10.1016/j.bbadis.2014.09.004.
Compliance with Ethical Standards
Conflict of interest
The authors declare that they have no conflict of interests to disclose.
Research Involving Human Participants and/or Animals
This article does not contain any studies with human participants performed by any of the authors. All institutional and national guidelines for the care and use of laboratory animals were followed and approved by “Federico II” University Ethical committee. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted.
Source of Funding
Progetti di rilevante interesse nazionale (PRIN)-Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR) 2009 (prot. 2009EL5WBP_001), “Società italiana di ipertensione arteriosa” (SIIA) 2012 (prot. and “Fondo di Ateneo per la ricerca di base” (FARB) 2011 (prot. 300397FRB14IACCA) to G.I.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editor-in-Chief Jennifer L. Hall oversaw the review of this article
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 34 kb)
Supplementary Figure 1
Effects of GRK2 on the regulation of BNP gene expression. Gene expression of BNP, a marker of hypertrophy together with ANF, was evaluated by real time PCR. GRK2 overexpression increased BNP gene expression and enhanced the response to PE. The overexpression of GRK2-DN inhibited BNP gene expression both basally and in response to PE (* p < 0.05 vs CTRL; # p < 0.05 vs PE). (JPEG 584 kb)
Supplementary Figure 2
Effects of GRK2 on angiotensin II-induced hypertrophy in vitro. To evaluate whether GRK2 inhibition was effective also in response to other hypertrophic stimuli, besides alpha-adrenergic activation, we performed a real time PCR to evaluate ANF gene expression in response to angiotensin II. The overexpression of GRK2 increased ANF gene expression and enhanced the response to angiotensin II. On the contrary, the overexpression of GRK2-DN inhibited ANF gene expression both basally and in response to angiotensin II (* p < 0.05 vs CTRL; # p < 0.05 vs angiotensin II). (JPEG 751 kb)
Supplementary Figure 3
Echocardiographic analysis of SHR rats. We report m-mode echo representative images of WKY, SHR and treated SHR groups. Y axis represents the depth (mm) and X-axis represents time (ms). (JPEG 1720 kb)
Supplementary Figure 4
Echocardiographic analysis of WKY rats. We report m-mode echo representative images of WKY, WKY treated with PE and WKY treated with PE and Ant-124. Y axis represents the depth (mm) and X-axis represents time (ms). (JPEG 3758 kb)
Rights and permissions
About this article

Cite this article
Sorriento, D., Santulli, G., Franco, A. et al. Integrating GRK2 and NFkappaB in the Pathophysiology of Cardiac Hypertrophy. J. of Cardiovasc. Trans. Res. 8, 493–502 (2015). https://doi.org/10.1007/s12265-015-9646-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12265-015-9646-0