
Abstract
Reprogramming of pig somatic cells to induced pluripotent stem cells provides a tremendous advance in the field of regenerative medicine since the pig represents an ideal large animal model for the preclinical testing of emerging cell therapies. However, the current generation of pig-induced pluripotent stem cells (piPSCs) require the use of time-consuming and laborious retroviral or lentiviral transduction approaches, in order to ectopically express the pluripotency-associated transcription factors Oct4, Sox2, Klf4 and c-Myc, in the presence of feeder cells. Here, we describe a simple method to produce piPSC with a single transfection of a CAG-driven polycistronic plasmid expressing Oct4, Sox2, Klf4, c-Myc and a green fluorescent protein (GFP) reporter gene, in gelatine-coated plates, with or without feeder cells. In our system, the derivation of piPSCs from adult pig ear fibroblasts on a gelatine coating showed a higher efficiency and rate of reprogramming when compared with three consecutive retroviral transductions of a similar polycistronic construct. Our piPSCs expressed the classical embryonic stem cell markers, exhibit a stable karyotype and formed teratomas. Moreover, we also developed a simple method to generate in vitro spontaneous beating cardiomiocyte-like cells from piPSCs. Overall, our preliminary results set the bases for the massive production of xeno-free and integration-free piPSCs and provide a powerful tool for the preclinical application of iPSC technology in a large animal setting.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The remarkable discovery of induced pluripotent stem (iPS) cells using exogenous factors has changed the future direction for stem cell research [1, 2]. The generation of embryonic stem-like cells by introducing only four factors: c-Myc, Klf4, Oct4 and Sox2 [1–4] or Lin 28, Nanog, Oct4 and Sox2 [5] into differentiated cells represented, therefore, a novel source of pluripotent stem cells. The initial establishment of iPS cells (iPSCs) held great potential for basic research, drug discovery and cell therapy approaches based on patient-specific iPS cells, which avoids immune rejection. However, human iPSC generation still presents some problems, among which a standout is the very low efficiency of colony formation [2]. In humans, some cell types, such as keratinocytes [6], neural stem cells [7], cord blood cells [8], or adipose stem cells [9], reprogram more efficiently than fibroblasts [1], suggesting that efforts should still be concentrated in order to find a highly susceptible and accessible tissue for clinical iPSCs generation. Until now, the constitutive retroviral and lentiviral vectors, commonly used for reprogramming, allowed efficient generation of iPS cells from somatic cells, but their permanent integration into the cell genome prohibits their use for eventual therapeutic applications, due to the risk of both insertional mutagenesis and, particularly, the reactivation of the reprogramming factors, which can lead to tumor formation [10]. In the last six years, several fancy but extremely low efficiency reprogramming strategies that avoid genomic integration have been developed, including the use of non-integrating adenoviruses [11, 12], plasmid transfection methods [13–17] and small chemical compounds that could substitute for the genetic factors [18].
On the other hand, iPSCs generation has been reported for mouse [1, 2], human [19], rat [20, 21] and monkey [22], suggesting that virtually any mammalian species can be used for iPS cell derivation. Mice are, and will probably continue to be, the unrivaled and easiest model to learn about the reprogramming machinery and to improve iPSCs methodology. However, the mouse model is rather unreliable to adapt for use in human clinical applications, considering the physiological and morphological differences between mice and human species. Moreover, the reduced life span, small size and low genetic variability in mice are handicaps for the evaluation of long-term effects of cell replacement and safety. Monkeys certainly represent the best alternative from a phylogenetic point of view, but ethical concerns and major difficulties in maintaining and breeding them impede their broader use.
On the contrary, the pig livestock farming has been adapted for thousands of years. In addition, the pig has a long-standing tradition as a meaningful model in many branches of medicine, notably for transplantation medicine, immunology and circulatory system surgery, because of its large morphological and functional affinity with man [23]. Nevertheless, isolation of fully competent and validated embryonic stem cells (ESCs) from these animals was still not achieved, despite years of maintained effort. Last year, three different publications reported, almost simultaneously, the successful derivation of induced pluripotent stem cells from swine [24–26]. These studies showed that pig somatic cells could be reprogrammed to iPS cells using a viral system expressing the four Yamanaka factors. We have recently described the generation of pig iPS cells (piPSCs) by retroviral transduction, showing that the obtained piPSCs displayed alkaline phosphatase activity, ESC surface markers, high telomerase activity and a normal karyotype (Oral Comunication in the Seventh International Symposium on Stem Cell Therapy and Cardiovascular Innovations, Nuria Montserrat). Embryoid body and teratoma formation assays indicated that they could also differentiate into cell types of the three embryonic germ layers.
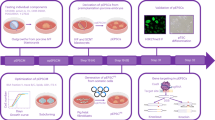
In the present work, we show the generation of piPSCs by transient expression of a single non-viral polycistronic vector in pig derived fibroblasts. We have developed a simple method, by means of a single transfection, to transiently co-express Oct4, Sox2, Klf4, c-Myc and the GFP reporter gene (pCAG-OSKMG). Following a protocol of a single transfection, we were able to generate piPSCs in approximately eight days. Furthermore, in order to mimic a therapeutic context, piPSCs have been generated in the absence of feeder cells, thus being closer to xeno-free conditions. These piPS cell lines express the typical set of pluripotency marker genes, posses a stable karyotype and are able to differentiate in vitro and in vivo into derivatives of the three embryonic germ layers. In addition, and for the first time, spontaneous beating cardiomyocyte-like cells were produced from our piPSCs after 15 days of differentiation in a specific differentiation media.
Materials and Methods
Fibroblast Isolation
Fibroblasts used in this study were derived from ear biopsies of a six-month-old white landrace × large white (pig) females. Cells were cultured in high glucose Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen), 10% fetal bovine serum (FBS; Invitrogen), GlutaMAX (200 mM; Invitrogen), penicillin–streptomycin (100 U·ml−1 penicillin and 100 μg·ml−1 streptomycin; Sigma). All the experiments were performed with passage 2 adult fibroblasts. Ear biopsies were kindly provided by M. Esteves (General Surgery Department, Universitary Hospital Vall d’Hebron, Barcelona).
Plasmid Constructions and Retroviral Production
The pMXs–Oct4–Sox2–Klf4–Myc–GFP (hence referred to as pMXs-OSKMG) vector generation was described previously in [8]. Single-factor retroviral plasmids consisting of pMSCV-based retroviral vectors expressing FLAG-tagged OCT4 (POU class 5 homeobox 1; abbreviated as Octamer-4), Sox2 (sex-determining region Y-box 2), Klf4 (Krueppel-like factor 4), or c-Myc (myelocytomatosis viral oncogene homolog) are available from Addgene (e.g., 20072, 20073, 20074 and 20075, respectively). Retroviruses were produced as previously reported [14, 27]. Briefly, 1.5 × 106 gp-Phoenix 293 cells were plated per 100 mm culture dish and transfected the following day. Per reaction (culture dish), 9 μg of plasmid, 27 μL of FuGENE 6 Transfection Reagent (Roche) and 1 mL OptiMEM (Gibco) were used following the manufacturer’s instructions. The transfection protocol consisted in complexing the OptiMEM and FuGENE 6 for five min at room temperature before adding the pMXs-OSKMG [14] and pCMV-VSV-G plasmids (Addgene, 8454). Next, the solution was kept for another 20 min at room temperature before adding it dropwise onto the plates containing the gp-Phoenix 293 cells, which were then incubated at 37°C, 5% CO2 overnight. The following day, the medium was replaced and the cells incubated at 32°C, 5% CO2 overnight. The transfection efficiency was checked using a Leica inverted fluorescent microscope and the corresponding LAS AF software.
Transduction of Porcine Fibroblast Cells
Pools of 80,000 passage 2 fibroblasts were infected three times at 12-h intervals with retroviruses carrying polycistronic vectors and centrifuged at 750 relative centrifugal force at 32°C for 45 min. One day after the last infection (T = 1), cells were trypsinized and plated onto iMEFs or gelatine 0.1% (Millipore) in human ES (hES)/mouse ES (mES) media (1:1). hES medium: knockout (KO)-DMEM (Invitrogen), KO serum replacement (20%, Invitrogen), GlutaMAX (1 mM; Invitrogen), penicillin–streptomycin (100 U·ml−1 penicillin and 100 mg·ml−1 streptomycin; Sigma), non-essential amino acid solution (100 μM; Invitrogen), 20% FBS (Invitrogen), 2-mercaptoethanol (100 μM; Invitrogen), recombinant human fibroblast growth factor (bFGF; PeproTech; 10 ng·ml−1). mES media: high glucose DMEM (Invitrogen), leukemia inhibitory factor (LIF; 1,000 U·ml−1; Chemicon), 15% FBS, GlutaMAX (1 mM), penicillin–streptomycin (100 U·ml−1 penicillin and 100 mg·ml−1 streptomycin), non-essential amino acids (100 μM), 2-mercaptoethanol (100 μM). When colonies appeared, typically four to seven days later, OSKMG cells were passaged onto iMEF and picked mechanically.
Plasmid Construction and Transfection Protocol
Pools of 80,000 passage 2 fibroblasts were seeded in six-well plates and transfected the next day by means of FuGENE 6 Transfection Reagent (Roche), according to the manufacturer’s instructions. Briefly, 3 μg of plasmid, 9 μl of FuGENE 6, and 0.3 ml of OptiMEM (Gibco) were used per reaction. The transfection protocol consisted in complexing the OptiMEM and the FuGENE 6 for five min at room temperature before adding the pCAG-OSKMG plasmid, a single CAG-driven polycistronic plasmid expressing Oct4, Sox2, Klf4, c-Myc and the GFP reporter gene [14]. The solution was kept for another 20 min at room temperature before adding it dropwise onto the plates containing the primary pig fibroblasts and then placed in 37°C, 5% CO2 incubator overnight.
Culture and Passaging of piPSCs Colonies
Individual colonies were picked manually using an Olympus R SZX12 stereomicroscope as well as strippers and stripper tips (MXL3-150 μm, MidAtlantic Diagnostics). piPSCs were cultured in a 1:1 solution of hES medium and mES medium.
Molecular Analysis of piPSC Lines
Integration of the transgenes was checked by polymerase chain reaction (PCR) using one primer pair spanning the OSKMG sequence as previously described [14].
Immunofluorescence Analysis and Alkaline Phosphatase Staining
Cells were grown on plastic cover slide chambers and fixed with 4% paraformaldehyde for 15 min at room temperature. The following antibodies were used: anti-GFP (GFP; AVES, 1:250), NANOG (Everest Biotech, 1:100), TRA-1-60 (MAB), stage-specific embryonic antigen 4 (Hybridoma Bank), neuron-specific class III beta-tubulin (Tuj1, Covance, 1:500), glial fibrillary acidic protein (GFAP; Dako, 1:1,000), forkhead box protein A2 (R&D System, 1:50), GATA 4 (Santa Cruz, 1:200), Nestin (Hybridoma Bank, Iowa, 1:200), smooth muscle actin (SMA; Sigma, 1:200), sex-determining region Y-box 9 (Sox-9, R&D Systems, 1:200), insulin (Chemicon, 1:250), chondroitin sulfate (CS; Sigma, 1:200). Images were taken using a Leica SP5 confocal microscope. Direct alkaline phosphatase (AP) activity was analyzed using AP blue membrane substrate solution kit (Sigma, AB0300) according to the manufacturer’s guidelines. Images were taken using a stereomicroscope equipped with a camera and the LAS AF software from Leica.
High-resolution, G-banded Karyotype
Giemsa-banded (G-banded) karyotype analysis was performed on 80% confluent cells growing on Matrigel (BD Systems Ltd). Cells were treated with colcemid at 20 ng/mL, followed by a 45-min incubation at 37°C. Then, after trypsinizing the cells into a single cell suspension, they were treated with Carnoy fixative at −20°C. Finally, the samples were analyzed with the software Cytovision (Applied Imaging).
In Vitro Differentiation
In vitro differentiation assay was performed by embryoid bodies (EBs) formation according to standard protocols [1, 6]. Briefly, colonies were detached from the iMEF feeder layer or gelatine-coated wells by manual dissection and transferred into differentiation medium (high glucose DMEM, 20% FBS, 2 mM GlutaMAX) in low attachment plates (Corning). After two to three days of culture, EBs were transferred onto adherent, gelatine-coated tissue-culture dishes and were cultured in differentiation medium supplemented with either 10 mM ascorbic acid (for induction of mesoderm/cardiomyocyte differentiation), differentiation medium (for endoderm differentiation). For ectoderm differentiation, EBs were transferred onto matrigel coated dishes supplemented with differentiation medium and 1 μM all-trans retinoic acid. For all the conditions, cells were finally fixed for immunofluorescence analysis 15 to 20 days later.
Teratoma Formation by piPSCs
piPSCs were manually picked and, following dispase treatment, they were injected into the skin or testes of non-obese diabetic-severe combined immunodeficiency (NOD-SCID) mice. Palpable tumors were observed, typically, one to two months after injection. Tumor samples were collected, generally, in two months and processed for paraffin embedding and hematoxylin and eosin staining following standard procedures.
Results
Generation of piPSC Lines
In order to generate piPSCs with a single transfection protocol, a polycistronic vector expressing the four reprogramming factors, Oct4, Sox2, Klf4, c-Myc and the GFP (pCAG-OSKMG) was used to transfect adult porcine fibroblasts (Fig. 1a). One day after plasmid transfection (T = 1), cells were transferred onto iMEF feeders or just gelatine-coated plates with hES:mES medium. Seven days after plasmid transfection (T = 7) compact colonies were visible. As expected, piPSCs colonies were comprised of cells exhibiting a high nucleus to cytoplasm ratio with prominent nucleoli, together with positive AP staining (Fig. 1a). We compared our single transfection protocol with an efficient retroviral transduction method, using a polycistronic retroviral vector expressing the four reprogramming factors Oct4, Sox2, Klf4, c-Myc and GFP (pMXs-OSKMG) (Fig. 1b). In this case, piPSCs colonies appeared 9 days after transferring the transduced cells onto iMEF or gelatine-coated plates (T = 12). Both technical approaches showed positive results by means of piPSC colonies generation. However, here we show that piPSC generation by means of a single transfection is not only possible but also that piPSC colonies appeared consistently sooner, when compared with the standard retroviral transduction method and, additionally, this was independent of the substrate used (iMEF or gelatine).

Timeline of adult pig fibroblast reprogramming onto irradiated mouse embryonic fibroblasts (iMEF). a A single CAG-driven polycistronic plasmid expressing Oct4, Sox2, Klf4, c-Myc and green fluorescent protein (GFP; pCAG-OSKMG) was used for the generation of piPSC lines by a single transfection method. One day before (T = −1; T = time) plasmid transfection, 80,000 pig fibroblasts were seeded in every six wells of a six-well plate. Next day (T = 1), cells were transferred onto iMEF or gelatine with hES/mES medium. Typical stem cell-like colonies were clearly visible at 4 days after transferring infected cells onto gelatine (T = 5). Seven days after plasmid transfection (T = 7), compact colonies were clearly visible. Representative phase contrast and fluorescence images of piPSC lines after 7 days (T = 7). Scale bar, 50 μm. Alkaline phosphatase (AP) staining of piPSC lines. Magnification, ×3.6. b pMXs-OSKMGFP polycistronic construct expressing POU domain class 5 transcription factor 1 (abbreviated as Octamer-4 (Oct4)), Sox2, Klf4, Myc and GFP genes linked by P2A peptide sequences was used for the generation of piPSC lines by retrovirus transduction. One day before (T = −1) viral transduction, 80,000 pig fibroblasts were seeded in every six wells of a six-well plate. Three days post-infection (T = 3) cells were transferred onto feeders or gelatine. Typical stem cell-like colonies were clearly visible at 5 days after transferring infected cells onto gelatine (T = 8). Representative phase contrast and fluorescence images of piPSC lines were taken at T = 12. Scale bar, 50 μm. AP staining of piPSC lines. Magnification, ×3.6
Expression of Pluripotency Genes in piPSC Lines
Then, we further characterized the piPSC generated by our single transfection protocol. From day seven in culture, tightly packed colonies with a morphology similar to human ES appeared, both with iMEF and gelatine substrates. Individual colonies were picked manually and several piPSC lines were established and maintained as described above (“Materials and methods”). We proceeded to characterize the piPSC lines in terms of pluripotency. Previous studies have reported the isolation of piPSCs based on cell morphology and immunostaining for the embryonic surface markers SSEA-3, SSEA4, TRA-1-60, and TRA-1–81 [24–26]. We also performed immunofluorescence analysis and showed that piPSC lines were positive for NANOG, and both surface markers SSEA4 and Tra-1-60 (Fig. 2a). Additionally, we could confirm that, under our culture conditions, the generated piPSC lines had a normal karyotype (Fig. 2b).

Expression of pluripotency markers, karyotype analysis and molecular characterization of piPSC lines obtained by a single transfection method. a Immunofluorescence analysis of the piPSC lines showed that they were positive for NANOG and both surface markers Tra-1-60 and SSEA4. Scale bars, 50 and 100 μm. b High-resolution, G-banded karyotype indicating a normal chromosomal content in piPSC lines. c Genomic DNA PCR analysis for transgene integration in piPSC lines demonstrated that Sox2-Klf4 transgenes were integrated in all the analyzed piPSC lines (numbers 1, 2, 3 and 4). As expected, pig fibroblasts were negative of any transgene integration
Next, the established piPSC lines were subjected to a PCR of genomic DNA to analyze the integration of the polycistronic construct. We used primers specially designed to amplify a specific region of the pCAG-OSKMG vector (Sox2-Klf4) and found that all clones analyzed had integrated the transgene in their DNA (Fig. 2c). In addition, piPSC lines maintained the expression of GFP with the passages, suggesting that the transgene was not adequately silenced. This observation has already been described by three other groups, which generated piPSC by means of retroviral or lentiviral transduction methods [24–26].
EBs formation and in vitro differentiation
We tested whether piPSC lines could form EBs in the absence of bFGF and LIF by culturing them on a non-adhesive substrate (Fig. 3a). All piPSC lines examined could form EBs under such conditions. When placed on gelatine-coated plates and cultured in the presence of mesoderm, ectoderm or endoderm inducing medias, EBs readily attached to the substrate and began to spread and differentiate (Fig. 3b). So far, the few works describing the generation of piPSCs have not studied, in depth, the differentiation of these cells into specific lineages. This is an important issue in order to investigate the extent with which these cells are able to produce differentiated cell types with a stable phenotype and appropriate functionality after transplantation into recipient pigs, and how the inadequate silencing of the transgene could affect this process. We developed a method to induce the differentiation of piPSCs into the cardiomyocyte lineage. Three-day EBs were transferred onto 0.1% gelatine-coated substrates in relatively high density (≈10 EBs per well were seeded on plastic cover slide chambers) with differentiation media without supplementation of ascorbic acid, also known as vitamin C, and maintained for two more days. From day 5 until days 15–20, differentiation medium containing 10 mM ascorbic acid was replaced every second to third day. Starting on day 6 of differentiation (1 day after plating), each EB outgrowth was examined daily for spontaneously beating areas. Under these differentiation conditions, we could observe focused beating cells within 15–20 days of culture (Electronic supplementary material, Mov. 1). Treatment with ascorbic acid produced an increase in spontaneous and rhythmic contractile activity on the different piPSC lines tested so far. This ascorbic acid effect on cardiac differentiation of piPSCs was not mimicked by other compounds such as the DNA methyltransferases inhibitor 5-azacytidine, which has been described to induce cardiomyocyte differentiation in P19 cells [28]. Spontaneously beating cells were analyzed by immunofluorescence for the expression of GATA4, SMA and vimentin, showing a characteristic phenotype that resembled cardiomyocyte cells (Fig. 3c).

In vitro differentiation of piPSCs. a All the characterized piPSC lines form EBs in the absence of bFGF and LIF. b When placed on gelatine-coated plates and cultured in the presence of differentiation media, EBs readily attached to the substrate and began to spread and differentiate. c After 15 to 20 days of mesoderm media supplementation (differentiation media with 10 mM ascorbic acid) clusters of cells showed spontaneous beating. piPSCs under these conditions express SMA (green), GATA4 (red) and vimentin (green). Scale bars, 50 and 250 μm
On the other hand, we also checked if all the generated clones readily differentiated into derivatives of the three embryonic germ layers in vitro. We observed that piPSCs acquired specific markers representing all three embryonic germ layers after being subjected to each specific differentiation protocol, detailed in the Material and Methods section. Under our culture conditions, piPSCs were able to specifically differentiate into endoderm, ectoderm or mesoderm fates. In addition, differentiated piPSC lines still maintained the expression of GFP, suggesting that although the transgenes are not adequately silenced, the differentiation process is not affected (Fig. 4).

Representative images of in vitro differentiation of several piPSC lines into the three primary embryonic germ cell layers. Endoderm, FOXA2. Ectoderm, GFAP. Mesoderm, sex-determining region Y-box 9 (Sox9). Notice that GFP indicates that the transgene is not silenced. Scale bar, 100 μm
The last step to evaluate the pluripotency of our piPSC lines is the in vivo differentiation into derivates of the three embryonic germ layers after their injection into immunocompromised NOD-SCID beige mice. After 8–12 weeks, the animals were sacrificed and the teratomas were collected. Conventional histology showed that piPSCs differentiated in vivo to endoderm (*), ectoderm (**) or mesoderm fates (***) (Fig. 5). Moreover, when teratomas were analyzed by immunofluorescence, we clearly showed that the tested piPSC lines formed complex teratomas that still retained GFP expression. We could clearly identify different complex structures positive for markers of: endoderm (FOXA2 and insulin), ectoderm (GFAP, nestin, and Tuj-1) and mesoderm (SMA, Sox-9 and CS) (Fig. 6). Teratoma formation provided evidence that during the generation and differentiation of piPSCs, transgene silencing is definitively not required, suggesting that these lines could be used for future transplantation assays in the pig.

In vivo differentiation of piPSCs. piPSCs were injected into the testis of immunocompromised NOD-SCID beige mice. After 12 weeks, teratomas were collected and analyzed by conventional histology. piPSCs differentiated in vivo to endoderm (asterisk), ectoderm (double asterisk) or mesoderm fates (triple asterisk)

In vivo differentiation of piPSCs. Teratomas were analyzed by immunofluorescence, showing that the different complex structures were positive for markers of endoderm (FOXA2, insulin), ectoderm (GFAP, nestin, and Tuj-1) and mesoderm (SMA, Sox-9 and CS). Scale bars, 25 and 50 μm
Discussion
The pig has a long-standing tradition as a meaningful model in many branches of medicine, notably for transplantation medicine, immunology and circulatory system surgery, because of its large morphological and functional affinity with man. Organ dimensions are largely similar and the extended life span (18–25 years) makes long-term experiments possible. Nowadays, insulin obtained from pig is used to treat diabetes and transgenic pigs lacking a major xenoantigen ((1–3)-galactosyltransferase) carry hopes for xenotransplantation [29, 30]. Pig heart valves and skin have already been transplanted in humans for decades. Considering the latest advances in cell replacement therapies and the current popularity of the stem cell research field, the establishment of pluripotent cells imposes upon the pig model.
Nevertheless, isolation of fully competent and validated ESCs from ungulates has proven impossible, despite years of maintained effort. Also, the use of somatic cell nuclear transfer, in order to achieve genetic manipulation, is laborious and inefficient [29, 30]. A number of reasons might explain the problems encountered, like the choice of the wrong stage during embryonic development or inappropriate culture and cell passage conditions. Last year, three independent groups reported, almost simultaneously, the successful derivation of induced pluripotent stem cells from swine by means of viral vector transduction on different pig somatic sources [24–26]. These studies demonstrated that pig somatic cells could be reprogrammed to iPS cells using a viral system expressing the four Yamanaka factors: Oct4, Sox2, Klf4 and c-Myc. Moreover, pig iPSCs displayed alkaline phosphatase activity, ESC surface markers, high telomerase activity and a normal karyotype. Embryoid body and teratoma formation assays indicated that they could also differentiate into cell types of all three embryonic germ layers. Although the generation of these piPSC lines represented a remarkable finding in the field of cell biology in these species, the permanent integration of constitutive retroviral and lentiviral vectors in the reported piPSC lines limits their use for eventual therapeutic applications, due to the risk of both insertional mutagenesis and particularly the reactivation of the reprogramming factors, leading to tumor formation. Thus, piPSCs derivation methods will need to be improved and many efforts will be directed to the use of non-integrating strategies. Here, following a non-viral delivery protocol of a single transfection, we could generate piPSCs in approximately seven days (Fig. 1a). These results are comparable to our findings using retroviral vectors and, surprisingly, the efficiency of piPSC generation using the single transfection method is similar to the efficiency of our retroviral protocol (Fig. 1b). Moreover, our piPSC lines express the typical set of pluripotency marker genes, have a stable karyotype and are able to differentiate in vitro and in vivo into derivatives of the three embryonic germ layers. Unfortunately, our approach did not allow the generation of transgene-free piPSC lines, but still represents a simple non-viral alternative to produce piPSCs in a shorter time frame. In addition, it avoids the time-consuming and labor-intensive task of producing and working with potentially harmful retroviral or lentiviral particles over-expressing known oncogenes, such as Klf-4 and c-Myc.
The recent development of iPSC technology has opened new perspectives in the field of regenerative medicine, even more so in the case of the pig, in which the derivation of pig ESC is still a challenge. In this sense, the generation of piPSC offers new possibilities in the use of the pig as an animal model for studying genetic diseases, engineering organs or developing cell transplantation therapies. In particular, the cardiovascular system of the pig is quite similar to that of humans and the pig is well accepted as a good model for studying cardiovascular pathologies, such as myocardial infarction. For this purpose, we aimed to generate cardiomyocyte precursors from piPSC and developed a reproducible method to obtain spontaneously beating cells from piPSC. We characterized the cardiomyocyte-like cells by immunofluorescence for specific markers such as GATA4 and SMA. Ongoing studies by our laboratory are trying to extensively characterize those cells in vitro and in vivo, in order to assess their potential for future regenerative purposes. Overall, and although some efforts will still be needed to achieve the generation of transgene-free piPSCs, the results presented here are promising and set up new tools for the study of cardiovascular pathologies, among others, in the pig model.
References
Takahashi, K., & Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell, 26(4), 663–676.
Maherali, N., Sridharan, R., Xie, W., Utikal, J., Eminli, S., Arnold, K., et al. (2007). Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell, 1, 55–70.
Okita, K., Ichisaka, T., & Yamanaka, S. (2007). Generation of germline-competent induced pluripotent stem cells. Nature, 448, 313–317.
Wernig, M., Meissner, A., Foreman, R., Brambrink, T., Ku, M., Hochedlinger, K., et al. (2007). In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature, 448, 318–324.
Yu, J., Vodyanik, M. A., Smuga-Otto, K., Antosiewicz-Bourget, J., Frane, J. L., Tian, S., et al. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science, 318, 1917–1920.
Aasen, T., Raya, A., Barrero, M. J., Garreta, E., Consiglio, A., Gonzalez, F., et al. (2008). Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nature Biotechnology, 26(11), 1276–1284.
Kim, J. B., et al. (2008). Pluripotent stem cells induced from adult neural stem cells by reprogramming with two factors. Nature, 454, 646–650.
Giorgetti, A., Montserrat, N., Aasen, T., Gonzalez, F., Rodríguez-Pizà, I., Vassena, R., et al. (2009). Generation of induced pluripotent stem cells from human cord blood using OCT4 and SOX2. Cell Stem Cell, 5(4), 353–357.
Sun, N., Panetta, N. J., Gupta, D. M., Wilson, K. D., Lee, A., Jia, F., et al. (2009). Feeder-free derivation of induced pluripotent stem cells from adult human adipose stem cells. Proceedings of the National Academy of Sciences of the United States of America, 106(37), 15720–5.
Nakagawa, M., Koyanagi, M., Tanabe, K., Takahashi, K., Ichisaka, T., Aoi, T., et al. (2008). Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nature Biotechnology, 26(1), 101–6.
Zhou, W., & Freed, C. R. (2009). Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells, 27(11), 2667–74.
Stadtfeld, M., Nagaya, M., Utikal, J., Weir, G., & Hochedlinger, K. (2008). Induced pluripotent stem cells generated without viral integration. Science, 322(5903), 945–9.
Okita, K., Nakagawa, M., Hyenjong, H., Ichisaka, T., & Yamanaka, S. (2008). Generation of mouse induced pluripotent stem cells without viral vectors. Science, 322, 949–953.
Gonzalez, F., Barragan Monasterio, M., Tiscornia, G., Montserrat Pulido, N., Vassena, R., Batlle Morera, L., et al. (2009). Generation of mouse-induced pluripotent stem cells by transient expression of a single nonviral polycistronic vector. Proceedings of the National Academy of Sciences of the United States of America, 106(22), 8918–8922.
Woltjen, K., Michael, I. P., Mohseni, P., Desai, R., Mileikovsky, M., Hämäläinen, R., et al. (2009). piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature, 458(7239), 766–770.
Yu, J., Hu, K., Smuga-Otto, K., Tian, S., Stewart, R., Slukvin, I. I., et al. (2009). Human induced pluripotent stem cells free of vector and transgene sequences. Science, 324(5928), 797–801.
Kaji, K., Norrby, K., Paca, A., Mileikovsky, M., Mohseni, P., & Woltjen, K. (2009). Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature, 458(7239), 771–775.
Huangfu, D., Maehr, R., Guo, W., Eijkelenboom, A., Snitow, M., Chen, A. E., et al. (2008). Induction of pluripotent stem cells by defined factors is greatly improved by smallmolecule compounds. Nature Biotechnology, 26, 795–797.
Park, I. H., Zhao, R., West, J. A., Yabuuchi, A., Huo, H., Ince, T. A., et al. (2008). Reprogramming of human somatic cells to pluripotency with defined factors. Nature, 451, 141–146.
Li, W., & Ding, S. (2010). Generation of novel rat and human pluripotent stem cells by reprogramming and chemical approaches. Methods in Molecular Biology, 636, 293–300.
Liao, J., Cui, C., Chen, S., Ren, J., Chen, J., Gao, Y., et al. (2009). Generation of induced pluripotent stem cell lines from adult rat cells. Cell Stem Cell, 4(1), 11–5.
Liu, H., Zhu, F., Yong, J., Zhang, P., Hou, P., Li, H., et al. (2008). Generation of induced pluripotent stem cells from adult rhesus monkey fibroblasts. Cell Stem Cell, 3, 587–590.
Kim, H.-I., Yu, J. E., Lee, S. Y., Sul, A. Y., Jang, M. S., Rashid, M. A., et al. (2009). The effect of composite pig islet-human endothelial cell grafts on the instant blood-mediated inflammatory reaction. Cell Transplantation, 18(1), 31–37.
Esteban, M. A., Xu, J., Yang, J., Peng, M., Qin, D., Li, W., et al. (2009). Generation of induced pluripotent stem cell lines from Tibetan miniature pig. The Journal of Biological Chemistry, 284(26), 17634–17640.
Ezashi, T., Telugu, B. P., Alexenko, A. P., Sachdev, S., Sinha, S., & Roberts, R. M. (2009). Derivation of induced pluripotent stem cells from pig somatic cells. Proceedings of the National Academy of Sciences of the United States of America, 106(27), 10993–10998.
Wu, Z., Chen, J., Ren, J., Bao, L., Liao, J., Cui, C., et al. (2009). Generation of pig induced pluripotent stem cells with a drug-inducible system. The Journal of Biological Chemistry, 1(1), 46–54.
Ory, D. S., Neugeboren, B. A., & Mulligan, R. C. (1996). A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G psuedotypes. Proceedings of the National Academy of Sciences of the United States of America, 93(21), 11400–11406.
Choi, S. C., Yoon, J., Shim, W. J., Ro, Y. M., & Lim, D. S. (2004). 5-azacytidine induces cardiac differentiation of P19 embryonic stem cells. Experimental & Molecular Medicine, 36(6), 515–23.
Brevini, T. A., Antonini, S., Cillo, F., Crestan, M., & Gandolfi, F. (2007). Porcine embryonic stem cells: facts, challenges and hopes. Theriogenology, 68(Suppl 1), S206–213.
Brevini, T. A., Antonini, S., Pennarossa, G., & Gandolfi, F. (2008). Recent progress in embryonic stem cell research and its application in domestic species. Reproduction in Domestic Animals, 43(Suppl. 2), 193–199.
Acknowledgements
We are grateful to Meritxell Carrió and Laetitia Casano for expert assistance with cell culture techniques, José Miguel Andrés Vaquero for assistance with flow cytometry, Lola Mulero Pérez, Cristina Pardo and Mercé Gaudes Martí for bioimaging assistance, Cristina Gómez and Cristina Morera for expert assistance in molecular biology techniques. NM was partially supported by Juan de la Cierva Program, EG was partially supported by Sara Borrell Program, AMCR supported by Fundação para a Ciência e Tecnologia. This work was partially supported by grants from MICINN, TERCEL, CIBER and Fundación Cellex.
Author information
Authors and Affiliations
Corresponding author
Additional information
Núria Montserrat and Elena Garreta Bahima contributed equally to this work
Electronic supplementary material
Below is the link to the electronic supplementary material.
(9.62 MB)
Rights and permissions
About this article
Cite this article
Montserrat, N., Bahima, E.G., Batlle, L. et al. Generation of Pig iPS Cells: A Model for Cell Therapy. J. of Cardiovasc. Trans. Res. 4, 121–130 (2011). https://doi.org/10.1007/s12265-010-9233-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12265-010-9233-3