
Abstract
MicroRNAs (miRNAs) are small and highly conserved non-coding RNA molecules that function to regulate gene expression. They play important roles in regulating cardiac physiological and pathological events such as hypertrophy, apoptosis, and heart failure. Induction of apoptosis in cardiomyocytes cannot be compensated by efficient cell proliferation, thereby leading to pathophysiological disorders. The miRNAs involved in cardiac apoptosis may provide a mechanism for the pathogenesis and treatment of heart diseases. This review summarizes the role of miRNAs in regulating cardiac apoptosis. In particular, it discusses the potential therapeutic approaches for apoptosis-related cardiac diseases by modulating miRNAs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
MicroRNAs (miRNAs) are a class of small non-coding RNAs that mediate posttranscriptional gene silencing. Recently, the work on miRNAs renovates our understanding about the gene regulation in cardiac physiology and pathology. The cardiac-specific knockout of Dicer, an RNAse III endonuclease critical for processing of pre-miRNAs into mature miRNAs, leads to rapidly progressive dilated cardiomyopathy, heart failure, and postnatal lethality [1, 2]. Knockout of an individual miRNA may result in pathological disorders. Targeted deletion of the muscle-specific miRNA, miR-1-2, reveals numerous functions in the heart including regulation of cardiac morphogenesis, electrical conduction, and cell-cycle control [3]. These results suggest that miRNAs play critical roles in maintaining normal cardiac structure and function. Cardiomyocyte apoptosis is related to cardiac disorders such as myocardial infarction, cardiomyopathy, cardiac hypertrophy, and anthracycline-induced cardiotoxicity [4–10]. The growing evidence demonstrates that miRNAs can regulate apoptosis [11–14]. This article reviews the role of miRNAs in regulating cardiac apoptosis. Specifically, it discusses the potential therapeutic approaches for apoptosis-related cardiac diseases by modulating miRNAs.
How do miRNAs Regulate Apoptosis?
Apoptosis can be initiated by a variety of miRNAs. miR-200a is able to target the E-cadherin and Wnt/β-catenin signaling pathways [15]. E2F1 is negatively regulated by miR-330, the latter induces apoptosis through E2F1-mediated suppression of Akt phosphorylation [16]. miR-122 modulates cyclin G1 silencing and increases sensitivity to doxorubicin challenge [17]. Upregulation of miR-23a-27a-24-2 cluster induces caspase-dependent and caspase-independent apoptosis [18]. Histone deacetylases are related to cell survival, and their expression can be repressed by miR-449a [19]. miR-101 and miR-29b exert their proapoptotic function via targeting Mcl-1 [20, 21]; the latter is a member of Bcl-2 family and can inhibit apoptosis [22, 23]. miR-34a inhibits silent information regulator 1 (SIRT1) expression leading to an increase in acetylated p53 and expression of p21 and p53 upregulated modulator of apoptosis [24]. These results suggest that miRNAs can provoke apoptosis.
Many miRNAs have been found to be able to inhibit apoptosis. miR-206 prevents apoptosis by targeting notch3 [25]. miR-17-92 cluster is a novel target for p53-mediated transcriptional repression under hypoxia, and overexpression of miR-17-92 cluster markedly inhibits hypoxia-induced apoptosis [26]. In addition, p53 is also regulated by miR-125b [27]. A summary of miRNAs functions in apoptosis is shown in Table 1.
Although the functions of the above miRNAs in apoptosis have been tested in other cell types, it can be speculated that they may have an impact in cardiomyocyte apoptosis. This is because their targets such as E2F1 and p53 have been proven to participate in the apoptotic program of cardiomyocytes [34–36].
miRNAs in Apoptosis-Related Cardiac Pathology
Cardiac hypertrophy is an early milestone of many heart diseases [37, 38] and associated with changes in gene expression [39]. It is related to apoptosis that plays a driving role in the transition from compensated hypertrophy to failure in the work-overloaded myocardium [7, 8, 40–42]. Recently, miRNAs are shown to regulate cardiac hypertrophy [43, 44]. Functional studies reveal that different miRNAs have distinct effects on cardiac hypertrophy. For example, inhibition of miR-133 causes significant cardiac hypertrophy [45]. In contrast, miR-208 is required for cardiomyocyte hypertrophy in response to stress and hypothyroidism [46, 47]. Cardiac-specific overexpression of miR-195 in mice results in severe cardiac hypertrophy [48]. Overexpression of miR-214 in the cardiomyocytes causes significant hypertrophy [48]. miR-1 negatively regulates the expression of hypertrophy-associated calmodulin and Mef2a genes [49]. Isoproterenol and aldosterone stimulate miR-23a expression. Knockdown of miR-23a could attenuate hypertrophy, suggesting that miR-23a is able to convey the hypertrophic signal. Nuclear factor of activated T cells can directly activate miR-23a expression through the transcriptional machinery. The muscle-specific ring finger protein 1, an anti-hypertrophic protein [50], is identified to be a target of miR-23a, and its translation could be suppressed by miR-23a [51]. Thus, it appears that miRNAs play multiple and essential roles in the regulation of cardiac hypertrophy. Future studies are required to elucidate whether miRNAs play a role for the transition from hypertrophy to apoptosis.
Heart failure is characterized by the occurrence of apoptosis [6, 52]. Dgcr8 controls microRNA biogenesis, and cardiomyocyte-specific deletion of dgcr8 leads to a fully penetrant phenotype that begins with left ventricular malfunction progressing to a dilated cardiomyopathy and premature lethality [53]. Intriguingly, miRNA expression patterns are distinct in two types of heart failure: idiopathic dilated cardiomyopathy and ischemic cardiomyopathy [54].
The excessive apoptosis has been identified in myocardial infarction [55, 56]. The growing evidence shows that miRNAs are related to acute myocardial infarction. In analyzing the expression patterns of miRNAs in noninfarcted and infarcted areas, 38 miRNAs are differentially expressed in infarcted areas, and 33 miRNAs are aberrantly expressed in the border areas [57]. It is of note that miR-21 expression is decreased in infarcted areas but is elevated in border areas. Enforced expression of miR-21 leads to a reduction in myocardial infarct size [57]. In response to ischemia/reperfusion, miR-320 expression is decreased in the hearts. Transgenic mice with cardiac-specific overexpression of miR-320 demonstrate an increased extent of apoptosis and infarction size in the hearts upon ischemia/reperfusion [33]. Acute myocardial infarction is related to the reduction of miR-29 [58]. A recent report shows that miR-1, miR-133a, miR-133b, and miR-208 are dysregulated in human myocardial infarction [59]. The broad involvement of miRNAs in apoptosis-related cardiac pathology necessitates the elucidation of the molecular mechanism by which they control apoptosis and the identification of their direct and indirect targets.
miRNAs Control Apoptotic Program of the Heart
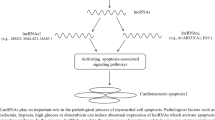
miRNAs have been characterized to regulate cardiac apoptotic program (Fig. 1). In response to ischemia/reperfusion, miR-320 expression is decreased in the heart. The functional study reveals that overexpression of miR-320 enhances apoptosis in cardiomyocytes, whereas knockdown of miR-320 can attenuate cell death upon ischemia/reperfusion. In searching for the targets of miR-320, heat-shock protein 20 (Hsp20) known as a cardioprotective protein, is proved to be a target for miR-320 in regulating apoptosis [33].

Summary of the apoptotic targets of microRNAs in the heart. See text for detailed explanations. Plus sign indicates that the final effect is the activation of apoptosis. Minus sign indicates that the final effect is the inhibition of apoptosis
miR-21 can regulate the ERK-MAP kinase signaling pathway in cardiac fibroblasts, which has impacts on global cardiac structure and function [60]. Programmed cell death 4 also is a direct target of miR-21 [29]. miR-199a inhibits hypoxia-inducible factor (Hif)-1α and its stabilization of p53 thereby reducing apoptosis. Knockdown of miR-199a during normoxia results in the upregulation of Hif-1α and SIRT1 and reproduces hypoxia preconditioning [32]. miR-1 participates in the activation of apoptosis by reducing the expression levels of heat shock protein-60 and heat shock protein-70, whereas miR-133 antagonizes apoptosis by repressing caspase-9 expression [28].
miRNAs inhibit apoptosis through multiple manners including the mitochondrial apoptotic pathway. Mitochondrial morphology is an important determinant of mitochondrial function [61]. Mitochondria constantly undergo fusion and fission that are necessary for the maintenance of organelle fidelity [62]. However, abnormal mitochondrial fusion and fission participate in the regulation of apoptosis. Mitochondrial fusion is able to inhibit apoptosis, while mitochondrial fission is involved in the initiation of apoptosis [63]. Mitochondrial fission is initiated by dynamin-related protein-1 (Drp1) [62, 64, 65]. miR-30 family members are able to inhibit mitochondrial fission and apoptosis by suppressing the expression of p53 that can upregulate Drp1 [30]. Taken together, this body of evidence demonstrates that miRNAs are able to regulate cardiac apoptotic machinery. It is expected that more miRNAs and their targets in the apoptotic program of cardiomyocytes will be delineated in the coming years.
miRNAs as the Therapeutic Targets for Apoptosis-Related Cardiac Diseases
Given the importance of miRNAs in cardiac physiology and pathology, can miRNAs be employed for the therapy of apoptosis-related heart diseases? Stem cells have been demonstrated to be a promising therapeutic approach for myocardial ischemic injury. Recently, there is a report showing that microRNAs can regulate the fate of stem cells. Ischemic preconditioning of bone marrow-derived mesenchymal stem cells with ischemia/re-oxygenation supports their survival under subsequent longer exposure to anoxia, and this effect is mediated by miR-210 that can target caspase-8-associated protein-2 [66].
miR-92a controls angiogenesis [67]. Enforced overexpression of miR-92a in endothelial cells blocks angiogenesis in vitro and in vivo. In mouse models of limb ischemia and myocardial infarction, systemic administration of a miR-92a antagomir leads to enhanced blood vessel growth and functional recovery of the damaged tissue. Thus, miR-92a may serve as a valuable therapeutic target in the setting of ischemic diseases [67].
To employ miRNAs as therapeutic targets, it is necessary to modulate the levels of endogenous miRNAs. To this end, the engineered oligonucleotides termed as “antagomirs” have been widely used as silencers of endogenous miRNAs. These chemically modified or cholesterol-conjugated antagomirs are quite stable and, therefore, can be efficiently delivered to the heart [45, 51] and liver [31]. Nevertheless, the effective delivery into target tissues remains a major hurdle for the applications of antagomirs as well as the synthetic miRNA duplexes [68]. For example, the systematic injection of antagomirs is incapable of silencing miRNAs in the central nervous system, and a local injection into the mouse cortex is required for efficiently targeting miRNAs [69]. It remains to be solved as to how the antagomirs can be maximally delivered to the heart. Finally, the in vivo safety should be well-defined in preclinical animal models.
Future Prospects
It has become quite clear that miRNAs regulate a diverse set of cardiovascular events including cardiac development, hypertrophy, and angiogenesis [70–74]. Especially in the last few years, the emerging evidence has shown that miRNAs play an important role in regulating apoptosis in the heart [75, 76]. Around 800 human miRNAs have been so far identified [12], but few of them have been studied in apoptosis in the heart. It is apparent that future studies are required to elucidate the role of each miRNA in cardiac apoptosis. A characteristic of miRNAs is that they may have multiple targets. It is thus necessary to explore the molecular targets of miRNAs in the cardiac apoptotic machinery. The crucial goals in coming years would be finding out the therapeutic approaches for apoptosis-related cardiac diseases by modulating miRNAs.
References
Chen, J. F., Murchison, E. P., Tang, R., Callis, T. E., Tatsuguchi, M., Deng, Z., et al. (2008). Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proceedings of the National Academy of Sciences of the United States of America, 105, 2111–2116.
da Costa Martins, P. A., Bourajjaj, M., Gladka, M., Kortland, M., van Oort, R. J., Pinto, Y. M., et al. (2008). Conditional dicer gene deletion in the postnatal myocardium provokes spontaneous cardiac remodeling. Circulation, 118, 1567–1576.
Zhao, Y., Ransom, J. F., Li, A., Vedantham, V., von Drehle, M., Muth, A. N., et al. (2007). Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell, 129, 303–317.
Kang, P. M., & Izumo, S. (2000). Apoptosis and heart failure: A critical review of the literature. Circulation Research, 86, 1107–1113.
Elsasser, A., Suzuki, K., & Schaper, J. (2000). Unresolved issues regarding the role of apoptosis in the pathogenesis of ischemic injury and heart failure. Journal of Molecular and Cellular Cardiology, 32, 711–724.
Barlucchi, L., Leri, A., Dostal, D. E., Fiordaliso, F., Tada, H., Hintze, T. H., et al. (2001). Canine ventricular myocytes possess a renin-angiotensin system that is upregulated with heart failure. Circulation Research, 88, 298–304.
Crow, M. T., Mani, K., Nam, Y. J., & Kitsis, R. N. (2004). The mitochondrial death pathway and cardiac myocyte apoptosis. Circulation Research, 95, 957–970.
Kitsis, R. N., & Mann, D. L. (2005). Apoptosis and the heart: A decade of progress. Journal of Molecular and Cellular Cardiology, 38, 1–2.
Siwik, D. A., Tzortzis, J. D., Pimental, D. R., Chang, D. L., Pagano, P. J., Singh, K., et al. (1999). Inhibition of copper-zinc superoxide dismutase induces cell growth, hypertrophic phenotype, and apoptosis in neonatal rat cardiac myocytes in vitro. Circulation Research, 85, 147–153.
Kwon, S. H., Pimentel, D. R., Remondino, A., Sawyer, D. B., & Colucci, W. S. (2003). H(2)O(2) regulates cardiac myocyte phenotype via concentration-dependent activation of distinct kinase pathways. Journal of Molecular and Cellular Cardiology, 35, 615–621.
Krichevsky, A. M., & Gabriely, G. (2009). miR-21: A small multi-faceted RNA. Journal of Cellular and Molecular Medicine, 13, 39–53.
Schickel, R., Boyerinas, B., Park, S. M., & Peter, M. E. (2008). MicroRNAs: Key players in the immune system, differentiation, tumorigenesis and cell death. Oncogene, 27, 5959–5974.
Mendell, J. T. (2008). miRiad roles for the miR-17-92 cluster in development and disease. Cell, 133, 217–222.
He, X., He, L., & Hannon, G. J. (2007). The guardian's little helper: microRNAs in the p53 tumor suppressor network. Cancer Research, 67, 11099–11101.
Saydam, O., Shen, Y., Wurdinger, T., Senol, O., Boke, E., James, M. F., et al. (2009). Downregulated microRNA-200a in meningiomas promotes tumor growth by reducing E-cadherin and activating the Wnt/beta-catenin signaling pathway. Molecular and Cellular Biology, 29, 5923–5940.
Lee, K. H., Chen, Y. L., Yeh, S. D., Hsiao, M., Lin, J. T., Goan, Y. G., et al. (2009). MicroRNA-330 acts as tumor suppressor and induces apoptosis of prostate cancer cells through E2F1-mediated suppression of Akt phosphorylation. Oncogene, 28, 3360–3370.
Fornari, F., Gramantieri, L., Giovannini, C., Veronese, A., Ferracin, M., Sabbioni, S., et al. (2009). MiR-122/cyclin G1 interaction modulates p53 activity and affects doxorubicin sensitivity of human hepatocarcinoma cells. Cancer Research, 69, 5761–5767.
Chhabra, R., Adlakha, Y. K., Hariharan, M., Scaria, V., & Saini, N. (2009). Upregulation of miR-23a-27a-24-2 cluster induces caspase-dependent and -independent apoptosis in human embryonic kidney cells. PLoS ONE, 4, e5848.
Noonan, E. J., Place, R. F., Pookot, D., Basak, S., Whitson, J. M., Hirata, H., et al. (2009). miR-449a targets HDAC-1 and induces growth arrest in prostate cancer. Oncogene, 28, 1714–1724.
Su, H., Yang, J. R., Xu, T., Huang, J., Xu, L., Yuan, Y., et al. (2009). MicroRNA-101, down-regulated in hepatocellular carcinoma, promotes apoptosis and suppresses tumorigenicity. Cancer Research, 69, 1135–1142.
Garzon, R., Heaphy, C. E., Havelange, V., Fabbri, M., Volinia, S., Tsao, T., et al. (2009). MicroRNA 29b functions in acute myeloid leukemia. Blood, 114(26), 5331–5341.
Zhou, P., Qian, L., Kozopas, K. M., & Craig, R. W. (1997). Mcl-1, a Bcl-2 family member, delays the death of hematopoietic cells under a variety of apoptosis-inducing conditions. Blood, 89, 630–643.
Dzhagalov, I., Dunkle, A., & He, Y. W. (2008). The anti-apoptotic Bcl-2 family member Mcl-1 promotes T lymphocyte survival at multiple stages. Journal of Immunology, 181, 521–528.
Yamakuchi, M., Ferlito, M., & Lowenstein, C. J. (2008). miR-34a repression of SIRT1 regulates apoptosis. Proceedings of the National Academy of Sciences of the United States of America, 105, 13421–13426.
Song, G., Zhang, Y., & Wang, L. (2009). MicroRNA-206 targets notch3, activates apoptosis, and inhibits tumor cell migration and focus formation. Journal of Biological Chemistry, 284, 31921–31927.
Yan, H. L., Xue, G., Mei, Q., Wang, Y. Z., Ding, F. X., Liu, M. F., et al. (2009). Repression of the miR-17-92 cluster by p53 has an important function in hypoxia-induced apoptosis. EMBO Journal, 28, 2719–2732.
Le, M. T., Teh, C., Shyh-Chang, N., Xie, H., Zhou, B., Korzh, V., et al. (2009). MicroRNA-125b is a novel negative regulator of p53. Genes and Development, 23, 862–876.
Xu, C., Lu, Y., Pan, Z., Chu, W., Luo, X., Lin, H., et al. (2007). The muscle-specific microRNAs miR-1 and miR-133 produce opposing effects on apoptosis by targeting HSP60, HSP70 and caspase-9 in cardiomyocytes. Journal of Cell Science, 120, 3045–3052.
Cheng, Y., Liu, X., Zhang, S., Lin, Y., Yang, J., & Zhang, C. (2009). MicroRNA-21 protects against the H(2)O(2)-induced injury on cardiac myocytes via its target gene PDCD4. Journal of Molecular and Cellular Cardiology, 47, 5–14.
Li, J., Donath, S., Li, Y., Qin, D., Prabhakar, B. S., & Li, P. (2010). miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet, 6, e1000795.
Esau, C., Davis, S., Murray, S. F., Yu, X. X., Pandey, S. K., Pear, M., et al. (2006). miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metabolism, 3, 87–98.
Rane, S., He, M., Sayed, D., Vashistha, H., Malhotra, A., Sadoshima, J., et al. (2009). Downregulation of miR-199a derepresses hypoxia-inducible factor-1alpha and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circulation Research, 104, 879–886.
Ren, X. P., Wu, J., Wang, X., Sartor, M. A., Qian, J., Jones, K., et al. (2009). MicroRNA-320 is involved in the regulation of cardiac ischemia/reperfusion injury by targeting heat-shock protein 20. Circulation, 119, 2357–2366.
Long, X., Boluyt, M. O., Hipolito, M. L., Lundberg, M. S., Zheng, J. S., O'Neill, L., et al. (1997). p53 and the hypoxia-induced apoptosis of cultured neonatal rat cardiac myocytes. Journal of Clinical Investigation, 99, 2635–2643.
Nam, Y. J., Mani, K., Wu, L., Peng, C. F., Calvert, J. W., Foo, R. S., et al. (2007). The apoptosis inhibitor ARC undergoes ubiquitin-proteasomal-mediated degradation in response to death stimuli: identification of a degradation-resistant mutant. Journal of Biological Chemistry, 282, 5522–5528.
Yurkova, N., Shaw, J., Blackie, K., Weidman, D., Jayas, R., Flynn, B., et al. (2008). The cell cycle factor E2F-1 activates Bnip3 and the intrinsic death pathway in ventricular myocytes. Circulation Research, 102, 472–479.
Hunter, J. J., & Chien, K. R. (1999). Signaling pathways for cardiac hypertrophy and failure. New England Journal of Medicine, 341, 1276–1283.
Frey, N., & Olson, E. N. (2003). Cardiac hypertrophy: the good, the bad, and the ugly. Annual Review of Physiology, 65, 45–79.
Clerk, A., Cullingford, T. E., Fuller, S. J., Giraldo, A., Markou, T., Pikkarainen, S., et al. (2007). Signaling pathways mediating cardiac myocyte gene expression in physiological and stress responses. Journal of Cellular Physiology, 212, 311–322.
Foo, R. S., Mani, K., & Kitsis, R. (2005). Death begets failure in the heart. Journal of Clinical Investigation, 115, 565–571.
Nadal-Ginard, B., Kajstura, J., Leri, A., & Anversa, P. (2003). Myocyte death, growth, and regeneration in cardiac hypertrophy and failure. Circulation Research, 92, 139–150.
Kass, D. A., Bronzwaer, J. G., & Paulus, W. J. (2004). What mechanisms underlie diastolic dysfunction in heart failure? Circulation Research, 94, 1533–1542.
van Rooij, E., & Olson, E. N. (2007). MicroRNAs: powerful new regulators of heart disease and provocative therapeutic targets. Journal of Clinical Investigation, 117, 2369–2376.
Callis, T. E., & Wang, D. Z. (2008). Taking microRNAs to heart. Trends in Molecular Medicine, 14, 254–260.
Care, A., Catalucci, D., Felicetti, F., Bonci, D., Addario, A., Gallo, P., et al. (2007). MicroRNA-133 controls cardiac hypertrophy. Nature Medicine, 13, 613–618.
van Rooij, E., Sutherland, L. B., Qi, X., Richardson, J. A., Hill, J., & Olson, E. N. (2007). Control of stress-dependent cardiac growth and gene expression by a microRNA. Science, 316, 575–579.
Callis, T. E., Pandya, K., Seok, H. Y., Tang, R. H., Tatsuguchi, M., Huang, Z. P., et al. (2009). MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. Journal of Clinical Investigation, 119, 2772–2786.
van Rooij, E., Sutherland, L. B., Liu, N., Williams, A. H., McAnally, J., Gerard, R. D., et al. (2006). A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proceedings of the National Academy of Sciences of the United States of America, 103, 18255–18260.
Ikeda, S., He, A., Kong, S. W., Lu, J., Bejar, R., Bodyak, N., et al. (2009). MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Molecular and Cellular Biology, 29, 2193–2204.
Arya, R., Kedar, V., Hwang, J. R., McDonough, H., Li, H. H., Taylor, J., et al. (2004). Muscle ring finger protein-1 inhibits PKC{epsilon} activation and prevents cardiomyocyte hypertrophy. Journal of Cell Biology, 167, 1147–1159.
Lin, Z., Murtaza, I., Wang, K., Jiao, J., Gao, J., & Li, P. F. (2009). miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proceedings of the National Academy of Sciences of the United States of America, 106, 12103–12108.
Ding, B., Abe, J., Wei, H., Huang, Q., Walsh, R. A., Molina, C. A., et al. (2005). Functional role of phosphodiesterase 3 in cardiomyocyte apoptosis: implication in heart failure. Circulation, 111, 2469–2476.
Rao, P. K., Toyama, Y., Chiang, H. R., Gupta, S., Bauer, M., Medvid, R., et al. (2009). Loss of cardiac microRNA-mediated regulation leads to dilated cardiomyopathy and heart failure. Circulation Research, 105, 585–594.
Sucharov, C., Bristow, M. R., & Port, J. D. (2008). miRNA expression in the failing human heart: Functional correlates. Journal of Molecular and Cellular Cardiology, 45, 185–192.
Shiomi, T., Tsutsui, H., Matsusaka, H., Murakami, K., Hayashidani, S., Ikeuchi, M., et al. (2004). Overexpression of glutathione peroxidase prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation, 109, 544–549.
Sun, M., Dawood, F., Wen, W. H., Chen, M., Dixon, I., Kirshenbaum, L. A., et al. (2004). Excessive tumor necrosis factor activation after infarction contributes to susceptibility of myocardial rupture and left ventricular dysfunction. Circulation, 110, 3221–3228.
Dong, S., Cheng, Y., Yang, J., Li, J., Liu, X., Wang, X., et al. (2009). MicroRNA expression signature and the role of microRNA-21 in the early phase of acute myocardial infarction. Journal of Biological Chemistry, 284, 29514–29525.
van Rooij, E., Sutherland, L. B., Thatcher, J. E., DiMaio, J. M., Naseem, R. H., Marshall, W. S., et al. (2008). Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proceedings of the National Academy of Sciences of the United States of America, 105, 13027–13032.
Bostjancic, E., Zidar, N., Stajer, D., & Glavac, D. (2009). MicroRNAs miR-1, miR-133a, miR-133b and miR-208 are dysregulated in human myocardial infarction. Cardiology, 115, 163–169.
Thum, T., Gross, C., Fiedler, J., Fischer, T., Kissler, S., Bussen, M., et al. (2008). MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature, 456, 980–984.
McBride, H. M., Neuspiel, M., & Wasiak, S. (2006). Mitochondria: More than just a powerhouse. Current Biology, 16, R551–R560.
Tanaka, A., & Youle, R. J. (2008). A chemical inhibitor of DRP1 uncouples mitochondrial fission and apoptosis. Molecular Cell, 29, 409–410.
Cassidy-Stone, A., Chipuk, J. E., Ingerman, E., Song, C., Yoo, C., Kuwana, T., et al. (2008). Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Develpmental Cell, 14, 193–204.
Bras, M., Yuste, V. J., Roue, G., Barbier, S., Sancho, P., Virely, C., et al. (2007). Drp1 mediates caspase-independent type III cell death in normal and leukemic cells. Molecular and Cellular Biology, 27, 7073–7088.
Wasiak, S., Zunino, R., & McBride, H. M. (2007). Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. Journal of Cell Biology, 177, 439–450.
Kim, H. W., Haider, H. K., Jiang, S., & Ashraf, M. (2009). Ischemic preconditioning augments survival of stem cells via MIR-210 expression by targeting caspase-8 associated protein 2. J Biol Chem, 284(48), 33161–33168.
Bonauer, A., Carmona, G., Iwasaki, M., Mione, M., Koyanagi, M., Fischer, A., et al. (2009). MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science, 324, 1710–1713.
Soifer, H. S., Rossi, J. J., & Saetrom, P. (2007). MicroRNAs in disease and potential therapeutic applications. Molecular Therapy, 15, 2070–2079.
Krutzfeldt, J., Kuwajima, S., Braich, R., Rajeev, K. G., Pena, J., Tuschl, T., et al. (2007). Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Research, 35, 2885–2892.
Morton, S. U., Scherz, P. J., Cordes, K. R., Ivey, K. N., Stainier, D. Y., & Srivastava, D. (2008). microRNA-138 modulates cardiac patterning during embryonic development. Proceedings of the National Academy of Sciences of the United States of America, 105, 17830–17835.
Cordes, K. R., & Srivastava, D. (2009). MicroRNA regulation of cardiovascular development. Circulation Research, 104, 724–732.
Cordes, K. R., Sheehy, N. T., White, M. P., Berry, E. C., Morton, S. U., Muth, A. N., et al. (2009). miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature, 460, 705–710.
Suarez, Y., Fernandez-Hernando, C., Pober, J. S., & Sessa, W. C. (2007). Dicer dependent microRNAs regulate gene expression and functions in human endothelial cells. Circulation Research, 100, 1164–1173.
Poliseno, L., Tuccoli, A., Mariani, L., Evangelista, M., Citti, L., Woods, K., et al. (2006). MicroRNAs modulate the angiogenic properties of HUVECs. Blood, 108, 3068–3071.
Barringhaus, K. G., & Zamore, P. D. (2009). MicroRNAs: regulating a change of heart. Circulation, 119, 2217–2224.
Latronico, M. V., & Condorelli, G. (2009). MicroRNAs and cardiac pathology. Nature Reviews Cardiology, 6, 419–429.
Acknowledgment
We apologize to all colleagues whose work could not be cited due to space limitations.