
Abstract
The new WHO 2016 classification of renal neoplasia encounters the new entity called “clear cell papillary renal cell carcinoma” (ccpRCC). The ccpRCC has been long included as a subtype of clear cell RCC histotype and it actually ranges from 2 to 9% in different routinely available cohort of renal carcinomas. Of important note, ccpRCC does not show any recurrences or metastases or lymph-node invasion and the outcome is always good. We reviewed twenty-four publications with available follow-up for patients (no. 362) affected by clear cell papillary RCCs/renal adenomatoid tumours and notably ccpRCC harbors an indolent clinical behavior after a mean of 38 months (3,5 years) of follow-up. This paper reviews the histological, molecular and clinical features characterizing ccpRCC, with the goal of focusing the knowledge of the benign fashion of this new tumour entity, supporting the idea of a new renal cell adenoma recruited morphologically from ex conventional clear cell RCC tumours.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Clear cell papillary RCC (ccpRCC) has been recently introduced as a new tumour entity by the 2016 WHO classification of renal neoplasia [1]. Overall, renal cancer comprises a heterogeneous group of malignancies with different morphological and immunohistochemical features, genetic aberrations, and peculiar clinical behavior. The 2016 WHO classification of renal tumours describes more than 40 different histological subtypes, being clear cell renal cell carcinoma (ccRCC) the most frequent subtype (accounting for about 70–85% of renal epithelial cancers), followed by papillary RCC and chromophobe RCC [1]. In 2013, the International Society of Urological Pathology (ISUP) added five new histological entities: tubolocystic RCC, acquired cystic disease-associated RCC, the microphthalmia transcription factor (MiT)-family translocation RCCs [in particular t(6;11) RCC], hereditary leiomyomatosis syndrome-associated RCC, and clear cell (tubulo)papillary RCC [2]. Initially described by Tickoo et al. [3] as a neoplasm occurring in end-stage renal disease (ESRD) setting, ccpRCC arises mainly in patients with no functional kidney impairments as a sporadic tumor [4,5,6,7,8]. Notably, clear cell papillary RCC has been reported without any tumor recurrences or metastases with the major limitation represented by the cumulative years of follow-up. For these reasons the term carcinoma has been maintained.
In this review we focused on ccpRCC at all clinico-pathological levels, with the goal to highlight the absence of recurrences or metastases reported, clustering ccpRCC in the group of renal cell adenoma.
Materials & Methods
A comprehensive literature search was conducted independently by two authors (C.C. and L.C.) from the PubMed database. The following keywords were used: clear cell papillary renal cell carcinoma, clear cell tubulo-papillary renal cell carcinoma, renal adenomatoid tumours of the kidney. We limited our research to articles written in English and we included only studies with available follow-up. Twenty-four articles were selected.
Clinical Presentation & Epidemiology
Clinically, the age at presentation of ccpRCC does not differ from that of RCCs (mean 60 years; ranging from 18 to 88 years), with no sex predilection [2, 5, 8]. The diagnosis generally follows the incidental finding of a renal mass in a completely asymptomatic patient.
Clear cell papillary RCC is considerably an under-recognized epidemiological entity despite it being a relative common renal neoplasm, accounting for 1.9 to 4.1% [9,10,11] of all epithelial renal cell carcinomas representing the fourth most frequent RCC histotype [12].
Overall, ccpRCC is not uncommon among small low-grade RCC tumors, it can be correctly recognized by its unique histomorphological features and confirmed by routinely immunohistochemical analysis, and it has an excellent clinical outcome following resection [13].
Among the manuscript we reported clinico-pathological data on ccpRCC and renal adenomatoid tumours of the kidney (RAT) being considered by the WHO 2016 two synonyms (Tables 1 and 2).
Pathologic Features
Macroscopic Features
Clear cell papillary RCC usually presents as a small unicentric, unilateral, clearly circumscribed and well-encapsulated mass located in the kidney cortex [2, 12]. On gross examination, tumor can be completely solid, or exhibit cystic changes ranging from 10% to 95% of the tumor mass [14,15,16]. The majority of ccpRCC reported in literature is in pathological stage T1 (generally less than 4 cm – T1a tumor), with few pT2 tumors, rarely pT3a [8, 9, 17].
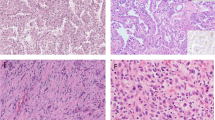
Macroscopic features are showed in Fig. 1a.

Clear cell-papillary RCC. Gross finding (a), haematoxylin and eosin staining (b), diffuse CK7 immunoexpression (c), strong high molecular weight cytokeratin (34bE12) (d), CAIX baso-lateral expression (e) and GATA-3 nuclear staining (f)ᅟ
Microscopic Characteristics
The most frequent architecture is the tubular-papillary, consisting of branching tubules and papillary projections [18]. Papillae can be rounded and small, or with branching contours. However, more complex architectures have been reported. Usually, a single layer of neoplastic cells with clear cell cytoplasm covers the papillae.
The tubular pattern may also predominates [19] and tubules show heterogeneous shapes and sizes [8].
Cystic component is present in almost all ccpRCC. The tumor structure can be mainly solid, with the cysts confined to the periphery of the mass, or predominantly cystic, with the peculiar papillae focally protruding within the cystic lumen.
Microscopic architecture is showed in Fig. 1b.
The cells lining the papillae and the cystic spaces are cuboidal, small to medium in size, typically with moderate to abundant optically clear cytoplasm. Characteristically, the nuclei, universally of low Fuhrman and low ISUP/WHO grade (small, round, with a regular border, often hyperchromatic, and uniform in shape, with inconspicuous nucleoli - grade 1 or 2), are typically localized in a linear fashion toward the luminal surface in the upper part of the cells and away from the basement membrane [5, 6, 13, 14, 20, 21].
Clear cell papillary RCC usually has a prominent stromal component made by fibrotic or collagenous tissue, with thin, fusiform nuclei, and small blood vessels finely scattered. When the stromal hyalinization is prominent can subvert and mask the entire epithelial component, making the histological diagnosis difficult [4].
Among aforementioned context, a tumour entity described by Michal Michal was initially reported and denominated renal adenomatoid tumour (RAT) [22].
Distinctive features of RAT include: 1) The angioleiomyomatous stroma, the amount of which may vary widely from less than 10% to involve the entire tumor, organized in bundles surrounding the entire tumor and supporting the epithelial tubular structures; 2) The epithelium characterized by secretory cells, with apical clear snouts, imparting to the glands the distinctive appearance of a “shark smile”. The basophilic nuclei present a low-grade (Fuhrman grades 1 and 2) and aligned in a basal position next to the basal membrane [23,24,25].
Morphological features along with immunohistochemical and molecular characteristics and the common benign biological behavior shared by ccpRCC and RAT lead to the hypothesis that these two originally distinct tumors represent actually morphological variants of the same cancer entity [18, 26]. The new WHO 2016 renal neoplasia classification have noted the inclusion of the RAT into the ccpRCC category after a consensus conference.
Immunohistochemical Features
The tumor shows diffuse and uniform immunoreactivity for CK7, with a co-expression of CA-IX [3, 6, 8, 11]. CK7 staining appears diffusely strong at cysts, papillae, and tubules/acini (Fig. 1c); CK7 positivity is also detectable, albeit of weaker intensity, in clear cell nests. The typical CAIX expression pattern in ccpRCC, with a diffuse membrane distribution lacking luminal borders staining (cup-shaped distribution) [3] (Fig. 1e), aids in the differential diagnosis with ccRCC [18]. Moreover, the majority of tumor cells strongly labels for high-molecular weight cytokeratin (34βE12) [19] (Fig. 1d), paired box gene 2 (PAX2), PAX8, vimentin, E-cadherin, β-catenin, c-MET, CK19, p27, p53. A strong immunostaining for HIF1 and GLUT-1, markers of HIF pathway, are characteristic of ccpRCC, and ccRCC, while contrasting with papillary-RCC [19].
The transcription factor GATA-3, involved in cancer development, tumor differentiation, epithelial-mesenchymal transition, stains positive in about one third of ccpRCC and is emerging as a specific marker [26]. Stains for CD10, RCC antigen, α-methylacyl-CoA racemase (AMACR), transcription factor E3 (TFE3), and translocation factor EB (TFEB) are generally negative [4, 19].
Therefore, the ccpRCC immunohistochemical phenotype (CK7+, CAIX+, AMACR−, 34βE12+, CD10- TFE3−) allows differentiating this tumor from other kidney cancer types with overlapping histomorphological features.
Molecular Features
Chromosomal Copy Number Alterations
Deletion of chromosome 3 and loss of the locus-specific subtelomeric chromosome 3p by fluorescence in situ hybridization [27] (and subsequent inactivation of several tumor suppressor genes, including VHL, BAP1, PBRM1 and SETD2], which represent a critical event of ccRCC carcinogenesis is typically absent in ccpRCC [4, 24].
Copy number variations of chromosomes 7, 17 and Y, are distinctive features of papillary RCCs but are absent in ccpRCC [4, 6].
Although it is debated, several analyses reported different chromosomal aberrations in ccpRCC, including trisomy of chromosomes 10 and 12 [28], monosomy of chromosomes 16, 17, and 20 by FISH [29].
Gene Mutations
Characteristically, ccpRCC, unlike ccRCC hystotype, do not harbor von Hippel-Lindau (VHL) gene mutations or VHL gene promoter methylation [19, 26]. Quantitative RT-PCR analysis reveals normal or even elevated VHL mRNA expression in ccpRCC compared to non-neoplastic controls, although in absence of VHL inactivating mutations or chromosome 3p losses. This finding could suggest the existence of alternative molecular mechanism leading HIF pathway up-regulation independent from VHL gene mutations, being ccpRCC positive for HIF [19].
Tumors that histologically resemble ccpRCC (defined as ccpRCC-like tumors) may occur in patients with VHL disease, but generally present an immunohistochemical (diffuse CD10 staining, negative CK7 labeling, and strong AMACR reactivity) and molecular profile (loss of 3p in about 80% by FISH - similar to ccRCC -, no chromosomes 7 or 17 aberrations) diverse from sporadic ccpRCC, implying a different pathogenesis [7, 29].
Using targeted next-generation sequencing (NGS) technology to sequence several common tumor-associated mutation ‘hot-spots’ in ccpRCC, Lawrie and Colleagues firstly identified few somatic mutations in ccpRCC [30] as Raspollini et al. [31]. Interestingly, Lawrie et al. observed mutations as follows: 1) mutations within intronic regions close to splice junction (comprising mutation in ERBB4, PTEN, and STK11 genes), whose functional significance is still unknown; 2) synonymous mutations, which do not directly modified the produced amino acid sequence; 3) non-synonymous mutations affecting the proto-oncogene MET (the T992I and N375S mutational variants) associated with epithelial-to-mesenchymal transition (EMT) [30]. Furthermore, the non-coding RNA expression analysis revealed a different expression of mature (entire miRnome), precursor (pre)-miRNAs, small nucleolar (sno)RNA and small Cajal body-specific (sca)RNAs between ccpRCC and ccRCC or papillary RCC subtypes [30]. Interestingly, and in contrast to other kidney cancer histotypes, the miRNA analysis showed the over-expression of all five members of the miR-200 family (miR-200a, miR200b, miR200c, miR-141, and miR-429 – regulators of EMT) in ccpRCC tumors, emphasizing the impaired (incomplete or blocked) process of EMT probably responsible for the indolent biological behavior of this entity [32].
The mRNA expression levels of eight genes typically altered in ccRCC and papillary RCC (AMACR, BMP and activin membrane-bound inhibitor homolog [BAMBI], CAIX, ceruloplasmin, nicotinamide N-methyltransferase [NNMT], schwannomin-interacting protein 1 [SCHIP1], solute carrier family 34 (sodium phosphate) member 2 [SLC34A2], and vimentin [VIM]) have been assessed by real-time (RT)-PCR among ccpRCC [30]. Although all eight genes were identified in ccpRCC, their levels of expression differed compared to other RCC histotypes. In particular, compared to papillary RCC, ccpRCC showed higher expression of CAIX, CP, NNMT, and VIM, lower AMACR, BAMBI, and SLC34A2 expression, and comparable levels of SCHIP1. Compared with ccRCC, ccpRCC expressed marginally less NNMT, but similar levels of the other genes [27].
All aforementioned details are sum up in Table 1, Figs. 2 and 3.

Genomic profiling of clear cell-papillary RCCs. Flat aCGH wide genomic profile, absence of cytogenetic deletion of chromosome 3p and absence of VHL gene mutation

Sum up of clinico-pathological characters of clear cell-papillary RCCs accoridng to WHO 2016 classification
Differential Diagnosis
A correct diagnosis of ccpRCC is of crucial importance [33] for the therapeutic management, the timing of follow-up and the impact on patient’s life. Three mainly relevant RCC tumor entities should be considered in the differential diagnosis: ccRCC, especially with low Fuhrman or low ISUP/WHO nucleolar grade, papillary RCC and multiocular cystic RCC with low malignant potential and rarely Xp11.2 translocation RCC. Clear cell RCCs, in contrast to ccpRCC, usually are positive for CD10, negative for CK7 and harbor VHL gene mutations/3p deletions.
The papillary architecture could raise problems in the differential diagnosis with papillary RCC. However, the clear cells are an uncommon finding in papillary RCCs. Moreover, the positive staining for AMACR, and the absence of reactivity for CAIX and HMWK in papillary RCCs help in challenging cases. Papillary RCCs (both type 1 and 2 subtypes) typically harbor genetic abnormalities such as trisomy of chromosomes 7 and 17, while type 2 papillary RCC frequently carries the Y chromosome loss which are lacking in ccpRCC [6, 19, 34]. MiT family translocation RCCs are rule out by the absence of cathepsin-k immunoexpression.
Clear cell papillary RCC and RAT has actually to be differentiated from conventional clear cell RCC with abundant fibroleiomyomatous stroma [35,36,37,38,39] and from a more recent distinct genomic tumor called TCEB1 mutated renal cell carcinoma [40].
Prognosis and Outcomes
All follow-up data and outcomes are summarized in Table 2 and Fig. 3.
Clear cell papillary RCC is characterized by an indolent clinical behavior, based on follow-up with a mean of 38 months reported (3,5 years) following 362 patients affected (see Table 2). In fact, no cases of local recurrence, lymph-node or distant metastases, and disease-related death have been described to date (see Table 2) irrespective of the clinic-pathological presentations (see Table 2). Moreover, the diagnosis of the vast majority of these tumors (small, well-circumscribed, unifocal masses) occurs in very early stage (stage I for almost all cases, rarely in stage II) (see Table 2). To further support the favorable clinical course related to ccpRCC, lympho-vascular invasions, tumor necrosis, and renal sinus infiltration - confirmed marks of negative prognostic value – have not been reported in association with ccpRCC. Recently, sarcomatoid transformation have been reported by Diolombi et al. [41]. Analogously to ccpRCC, RAT cases present a benign biological behavior, with no local/distant recurrence after surgery (see Table 2). The benign clinical course of ccpRCC/RAT tumors could biologically be justified by the activation of specific signaling pathways associated with good prognosis.
It is therefore not surprising that the debate on the most appropriate terminology to use for labeling this renal cancer subtype is still open. If the benign clinical behavior of this entity will be unequivocally confirmed, it would be conceivable to definitively abandon the misleading term of “carcinoma”? Carcinoma is an epithelial neoplastic lesion with the potential behavior to metastasize. Adenoma is an epithelial neoplastic lesion with absence of aforementioned capacity.
Up to date the term “clear cell papillary adenoma” could be replaced to these tumors as all reported evidenced benign appearance, no one tumor reported showed M1 staging along many years of follow-up, with no N1+ findings. It is not just mere semantics, but it would be of great importance especially 1) to reduce the patients psychological impact related to the diagnosis of a malignant neoplasia, 2) to avoid close and invasive follow-up, 3) to better guide therapeutic management of small renal tumors, 4) to avoid bias in the cohort of ccRCC used as control in studies focusing on prognostication, 5) to promote conservative surgery where possible or a conservative approach by active surveillance in high-risk surgical patients (single kidney or renal chronic failure patients).
Conclusion
The new tumor entity denominated “clear cell-papillary renal cell carcinoma/RAT” according to the WHO 2016 new classification, have the clinical characters of a renal cell adenoma as does harbor a benign outcome. The incidence range from 2 to 9% of consecutive renal cell neoplasia and the knowledge of aforementioned clinic-pathological information open new approaches (minimal surgical approach such as enucleation, active surveillance or minimal follow-up and appropriate identification with immunophenotypical and molecular analysis on tissue either whole tumour or biopsy) to patients affected by clear cell-papillary renal cell carcinoma/RAT neoplasms. Our review does highlight important information for oncologists such as the absence of any recurrence or any metastases for patients.
References
Moch H, Humphrey PA, Ulbright TM, Reuter VE (2016) WHO classification of Tumours of the urinary system and male genital organs (IARC WHO classification of Tumours). IARC, Lyon
Srigley JR, Delahunt B, Eble JN, Egevad L, Epstein JI, Grignon D, Hes O, Moch H, Montironi R, Tickoo SK, Zhou M, Argani P (2013) The International Society of Urological Pathology (ISUP) Vancouver classification of renal Neoplasia. Am J Surg Pathol 37(10):1469–1489. doi:10.1097/PAS.0b013e318299f2d1
Tickoo SK, dePeralta-Venturina MN, Harik LR, Worcester HD, Salama ME, Young AN, Moch H, Amin MB (2006) Spectrum of epithelial neoplasms in end-stage renal disease: an experience from 66 tumor-bearing kidneys with emphasis on histologic patterns distinct from those in sporadic adult renal neoplasia. Am J Surg Pathol 30(2):141–153
Aron M, Chang E, Herrera L, Hes O, Hirsch MS, Comperat E, Camparo P, Rao P, Picken M, Michal M, Montironi R, Tamboli P, Monzon F, Amin MB (2015) Clear cell-papillary renal cell carcinoma of the kidney not associated with end-stage renal disease: clinicopathologic correlation with expanded immunophenotypic and molecular characterization of a large cohort with emphasis on relationship with renal angiomyoadenomatous tumor. Am J Surg Pathol 39(7):873–888. doi:10.1097/PAS.0000000000000446
Adam J, Couturier J, Molinie V, Vieillefond A, Sibony M (2011) Clear-cell papillary renal cell carcinoma: 24 cases of a distinct low-grade renal tumour and a comparative genomic hybridization array study of seven cases. Histopathology 58(7):1064–1071. doi:10.1111/j.1365-2559.2011.03857.x
Gobbo S, Eble JN, Grignon DJ, Martignoni G, MacLennan GT, Shah RB, Zhang S, Brunelli M, Cheng L (2008) Clear cell papillary renal cell carcinoma: a distinct histopathologic and molecular genetic entity. Am J Surg Pathol 32(8):1239–1245. doi:10.1097/PAS.0b013e318164bcbb
Williamson SR, Zhang S, Eble JN, Grignon DJ, Martignoni G, Brunelli M, Wang M, Gobbo S, Baldridge LA, Cheng L (2013) Clear cell papillary renal cell carcinoma-like tumors in patients with von Hippel-Lindau disease are unrelated to sporadic clear cell papillary renal cell carcinoma. Am J Surg Pathol 37(8):1131–1139. doi:10.1097/PAS.0b013e318282dab8
Aydin H, Chen L, Cheng L, Vaziri S, He H, Ganapathi R, Delahunt B, Magi-Galluzzi C, Zhou M (2010) Clear cell tubulopapillary renal cell carcinoma: a study of 36 distinctive low-grade epithelial tumors of the kidney. Am J Surg Pathol 34(11):1608–1621. doi:10.1097/PAS.0b013e3181f2ee0b
Park JH, Lee C, Suh JH, Moon KC (2012) Clear cell papillary renal cell carcinoma: a report of 15 cases including three cases of concurrent other-type renal cell carcinomas. Korean J of Pathol 46(6):541–547. doi:10.4132/Korean JPathol.2012.46.6.541
Williamson SR, Eble JN, Cheng L, Grignon DJ (2013) Clear cell papillary renal cell carcinoma: differential diagnosis and extended immunohistochemical profile. Mod Pathol: Official J of the United States Canadian Acad Pathol Inc 26(5):697–708. doi:10.1038/modpathol.2012.204
Mai KT, Kohler DM, Belanger EC, Robertson SJ, Wang D (2008) Sporadic clear cell renal cell carcinoma with diffuse cytokeratin 7 immunoreactivity. Pathology 40(5):481–486. doi:10.1080/00313020802197962
Zhou H, Zheng S, Truong LD, Ro JY, Ayala AG, Shen SS (2014) Clear cell papillary renal cell carcinoma is the fourth most common histologic type of renal cell carcinoma in 290 consecutive nephrectomies for renal cell carcinoma. Hum Pathol 45(1):59–64. doi:10.1016/j.humpath.2013.08.004
Gill S, Kauffman EC, Kandel S, George S, Schwaab T, Xu B (2015) Incidence of clear cell papillary renal cell carcinoma in low-grade renal cell carcinoma cases: a 12-year retrospective Clinicopathologic study from a single cancer center. Int J Surg Pathol. doi:10.1177/1066896915613432
Alexiev BA, Thomas C, Zou YS (2014) Clear cell papillary renal cell carcinoma with angiomyomatous stroma: a histological, immunohistochemical, and fluorescence in situ hybridization study. Virchows Archiv: Intern J Pathol 464(6):709–716. doi:10.1007/s00428-014-1581-y
Bhatnagar R, Alexiev BA (2012) Renal-cell carcinomas in end-stage kidneys: a clinicopathological study with emphasis on clear-cell papillary renal-cell carcinoma and acquired cystic kidney disease-associated carcinoma. Int J Surg Pathol 20(1):19–28. doi:10.1177/1066896911414273
Alexiev BA, Drachenberg CB (2014) Clear cell papillary renal cell carcinoma: incidence, morphological features, immunohistochemical profile, and biologic behavior: a single institution study. Pathol Res Pract 210(4):234–241. doi:10.1016/j.prp.2013.12.009
Shi SS, Shen Q, Xia QY, Tu P, Shi QL, Zhou XJ, Rao Q (2013) Clear cell papillary renal cell carcinoma: a clinicopathological study emphasizing ultrastructural features and cytogenetic heterogeneity. Int J Clin Exp Pathol 6(12):2936–2942
Behdad A, Monzon F, Hes O, Michal M, Comperat E, Camparo P, Rao P, Picken M, Montironi R, Annaiah C, Herrera L, Arora K, Westfall DE, Tamboli P, Amin M (2011) Relationship between sporadic clear cell-papillary renal cell carcinoma (CP-RCC) and renal angiomyoadenomatous tumor (RAT) of the kidney: analysis by virtual karyotyping, fluorescent in situ analysis and immunohistochemistry (IHC). Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc 24(Suppl):179A
Rohan SM, Xiao Y, Liang Y, Dudas ME, Al-Ahmadie HA, Fine SW, Gopalan A, Reuter VE, Rosenblum MK, Russo P, Tickoo SK (2011) Clear-cell papillary renal cell carcinoma: molecular and immunohistochemical analysis with emphasis on the von Hippel-Lindau gene and hypoxia-inducible factor pathway-related proteins. Mod Pathol: Official J of the United States Canadian Acad Pathol Inc 24(9):1207–1220. doi:10.1038/modpathol.2011.80
Leroy X, Camparo P, Gnemmi V, Aubert S, Flamand V, Roupret M, Fantoni JC, Comperat E (2014) Clear cell papillary renal cell carcinoma is an indolent and low-grade neoplasm with overexpression of cyclin-D1. Histopathology 64(7):1032–1036. doi:10.1111/his.12359
Yan WX, Cao WR, Zhao J, Zhang W, Wang XL, Yuan Q, Dang SQ (2015) Clear cell papillary renal cell carcinoma: a clinicopathologic analysis of 6 cases. Int J Clin Exp Pathol 8(5):4595–4599
Michal M, Hes O, Havlicek F (2000) Benign renal angiomyoadenomatous tumor: a previously unreported renal tumor. Ann Diagn Pathol 4(5):311–315. doi:10.1053/adpa.2000.17890
Singh C, Kendi AT, Manivel JC, Pambuccian SE (2012) Renal angiomyoadenomatous tumor. Ann Diagn Pathol 16(6):470–476. doi:10.1016/j.anndiagpath.2012.01.006
Kuroda N, Michal M, Hes O, Taguchi T, Tominaga A, Mizobuchi K, Ohe C, Sakaida N, Uemura Y, Shuin T, Lee GH (2009) Renal angiomyoadenomatous tumor: fluorescence in situ hybridization. Pathol Int 59(9):689–691. doi:10.1111/j.1440-1827.2009.02429.x
Michal M, Hes O, Nemcova J, Sima R, Kuroda N, Bulimbasic S, Franco M, Sakaida N, Danis D, Kazakov DV, Ohe C, Hora M (2009) Renal angiomyoadenomatous tumor: morphologic, immunohistochemical, and molecular genetic study of a distinct entity. Virchows Archiv: Intern J Pathol 454(1):89–99. doi:10.1007/s00428-008-0697-3
Deml KF, Schildhaus HU, Comperat E, von Teichman A, Storz M, Schraml P, Bonventre JV, Fend F, Fleige B, Nerlich A, Gabbert HE, GaBler N, Grobholz R, Hailemariam S, Hinze R, Knuchel R, Lhermitte B, Nesi G, Rudiger T, Sauter G, Moch H (2015) Clear cell papillary renal cell carcinoma and renal angiomyoadenomatous tumor: two variants of a morphologic, immunohistochemical, and genetic distinct entity of renal cell carcinoma. Am J Surg Pathol 39(7):889–901. doi:10.1097/PAS.0000000000000456
Fisher KE, Yin-Goen Q, Alexis D, Sirintrapun JS, Harrison W, Benjamin Isett R, Rossi MR, Moreno CS, Young AN, Osunkoya AO (2014) Gene expression profiling of clear cell papillary renal cell carcinoma: comparison with clear cell renal cell carcinoma and papillary renal cell carcinoma. Mod Pathol 27(2):222–230. doi:10.1038/modpathol.2013.140
Wolfe A, Dobin SM, Grossmann P, Michal M, Donner LR (2011) Clonal trisomies 7,10 and 12, normal 3p and absence of VHL gene mutation in a clear cell tubulopapillary carcinoma of the kidney. Virchows Archiv: Intern J Pathol 459(4):457–463. doi:10.1007/s00428-011-1137-3
Rao P, Monzon F, Jonasch E, Matin SF, Tamboli P (2014) Clear cell papillary renal cell carcinoma in patients with von Hippel-Lindau syndrome--clinicopathological features and comparative genomic analysis of 3 cases. Hum Pathol 45(9):1966–1972. doi:10.1016/j.humpath.2014.06.004
Lawrie CH, Larrea E, Larrinaga G, Goicoechea I, Arestin M, Fernandez-Mercado M, Hes O, Caceres F, Manterola L, Lopez JI (2014) Targeted next-generation sequencing and non-coding RNA expression analysis of clear cell papillary renal cell carcinoma suggests distinct pathological mechanisms from other renal tumour subtypes. J Pathol 232(1):32–42. doi:10.1002/path.4296
Raspollini MR, Castiglione F, Cheng L, Montironi R, Lopez-Beltran A (2016) Genetic mutations in accordance with a low malignant potential tumour are not demonstrated in clear cell papillary renal cell carcinoma. J Clin Pathol. doi:10.1136/jclinpath-2015-203565
Munari E, Marchionni L, Chitre A, Hayashi M, Martignoni G, Brunelli M, Gobbo S, Argani P, Allaf M, Hoque MO, Netto GJ (2014) Clear cell papillary renal cell carcinoma: micro-RNA expression profiling and comparison with clear cell renal cell carcinoma and papillary renal cell carcinoma. Hum Pathol 45(6):1130–1138. doi:10.1016/j.humpath.2014.01.013
Dhakal HP, McKenney JK, Khor LY, Reynolds JP, Magi-Galluzzi C, Przybycin CG (2016) Renal neoplasms with overlapping features of clear cell renal cell carcinoma and clear cell papillary renal cell carcinoma: a Clinicopathologic study of 37 cases from a single institution. Am J Surg Pathol 40(2):141–154. doi:10.1097/PAS.0000000000000583
Gobbo S, Eble JN, Maclennan GT, Grignon DJ, Shah RB, Zhang S, Martignoni G, Brunelli M, Cheng L (2008) Renal cell carcinomas with papillary architecture and clear cell components: the utility of immunohistochemical and cytogenetical analyses in differential diagnosis. Am J Surg Pathol 32(12):1780–1786. doi:10.1097/PAS.0b013e31818649ed
Kuhn E, De Anda J, Manoni S, Netto G, Rosai J (2006) Renal cell carcinoma associated with prominent angioleiomyoma-like proliferation: report of 5 cases and review of the literature. Am J Surg Pathol 30(11):1372–1381. doi:10.1097/01.pas.0000213277.45715.82
Shannon BA, Cohen RJ, Segal A, Baker EG, Murch AR (2009) Clear cell renal cell carcinoma with smooth muscle stroma. Hum Pathol 40(3):425–429. doi:10.1016/j.humpath.2008.05.021
Iczkowski KA, Shanks JH, Burdge AH, Cheng L (2013) Renal cell carcinoma with clear cells, smooth muscle stroma, and negative for 3p deletion: a variant of renal angiomyoadenomatous tumour? A case report. Histopathology 62(3):522–524. doi:10.1111/his.12040
Martignoni G, Brunelli M, Segala D, Gobbo S, Borze I, Atanesyan L, Savola S, Barzon L, Masi G, Tardanico R, Zhang S, Eble JN, Chilosi M, Bohling T, Cheng L, Delahunt B, Knuutila S (2014) Renal cell carcinoma with smooth muscle stroma lacks chromosome 3p and VHL alterations. Mod Pathol: Official J of the United States Canadian Acad Pathol Inc 27(5):765–774. doi:10.1038/modpathol.2013.180
Canzonieri V, Volpe R, Gloghini A, Carbone A, Merlo A (1993) Mixed renal tumor with carcinomatous and fibroleiomyomatous components, associated with angiomyolipoma in the same kidney. Pathol Res Pract 189(8):951–956; discussion 957-959. doi:10.1016/S0344-0338(11)81110-6
Hakimi AA, Tickoo SK, Jacobsen A, Sarungbam J, Sfakianos JP, Sato Y, Morikawa T, Kume H, Fukayama M, Homma Y, Chen YB, Sankin AI, Mano R, Coleman JA, Russo P, Ogawa S, Sander C, Hsieh JJ, Reuter VE (2015) TCEB1-mutated renal cell carcinoma: a distinct genomic and morphological subtype. Mod Pathol: Official J of the United States Canadian Acad Pathol Inc 28(6):845–853. doi:10.1038/modpathol.2015.6
Diolombi ML, Cheng L, Argani P, Epstein JI (2015) Do clear cell papillary renal cell carcinomas have malignant potential? Am J Surg Pathol 39(12):1621–1634. doi:10.1097/PAS.0000000000000513
Acknowledgements
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Francesco Massari and Chiara Ciccarese co-authored as first.
Guido Martignoni and Matteo Brunelli are seniorship coauthor.
Rights and permissions
About this article

Cite this article
Massari, F., Ciccarese, C., Hes, O. et al. The Tumor Entity Denominated “clear cell-papillary renal cell carcinoma” According to the WHO 2016 new Classification, have the Clinical Characters of a Renal Cell Adenoma as does Harbor a Benign Outcome. Pathol. Oncol. Res. 24, 447–456 (2018). https://doi.org/10.1007/s12253-017-0271-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12253-017-0271-x