
Abstract
Monitoring of non small cell lung cancer (NSCLC) patients on afatinib after acquired resistance to 1st generation tyrosine kinase inhibitors is important. Circulating tumor DNA (ctDNA) offers an attractive means other than conventional tissue biopsy to characterize real time dynamic changes of the disease. In our study, we aim to ascertain the clinical value for ctDNA monitoring of NSCLC patients with acquired resistance for afatinib treatment. 200 patients positive for the activating epithermal growth factor receptor (EGFR) mutations were recruited for the study. Baseline molecular profiling for L858R, Exon 19 deletion and T790M were performed. Thereafter, serial blood samples were taken and patients were assessed by overall survival (OS) to determine the usefulness of ctDNA monitoring. At baseline, matched tumor biopsy and ctDNA analysis had a concordance agreement of 93.5% for L858R and exon 19 deletion. We also determined that a large proportion of patients had the drug resistance mutation T790M prior to starting afatinib and these patients were linked to a worse survival outcome. For patients that registered a drop in ctDNA levels after afatinib was administered, we observed that their survival outcome was more favorable (hazard ratio 1.56, (95% CI 1.04 to 2.43). ctDNA levels were mostly elevated after the 3rd sampling cycle. Our results suggest that ctDNA can be used to predict the clinical benefits of afatinib treatment. Pre and post blood sampling aids to identify patient groups that may benefit most from the treatment and ctDNA is relatively sensitive to address the dynamic changes of the disease at the molecular level.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tyrosine kinase inhibitors (TKIs) are the recommended first line treatments for non small cell lung cancer (NSCLC) patients with sensitizing EGFR mutations. These mutations are generally found in exons 18 to 21 of the TK domain [1, 2] and increase the kinase activity of the receptor. The mutations include L858R and exon 19 deletion that accounts for more than 90% of all activating EGFR mutations [3]. These prevalent EGFR mutations are well characterized, especially their clinical responses to TKI gefitinib and erlotinib [4, 5]. The mechanism of action involves competing with adenosine triphosphate (ATP) at the active site of the EGFR kinase[6, 7]. Early clinical trials using both gefitinib and erlotinib are encouraging, with patients showing improvement in both progression free and overall survival outcome[8, 9]. Despite the promising results, disease progression is inevitable for these patients due to the development of secondary drug resistance mechanisms [10, 11].
The second generation EGFR-TKI afatinib is an irreversible ErbB family blocker with demonstrated preclinical activity in numerous EGFR-mutant cell lines including T790M [12, 13]. This secondary point mutation (T790M) that substitutes methionine for threonine is present in more than 50% of patients who received an EGFR TKI [10, 14]. The recurrent mutation T790M is attributed to patients who developed acquired resistance after being treated with gefitinib or erlotinib. In a phase II trial by Katakami et al., afatinib was shown to possess notable drug efficacy in NSCLC patients who had received third- or fourth-line treatment with significant improvement in progression free survival of 11.1 months [15]. Afatinib is important and can play a significant role in addressing the issue of drug resistance.
Our study aims to prospectively analyze the treatment effectiveness of afatinib on patients who developed drug resistance using circulating tumor DNA (ctDNA) from peripheral blood. Analysis of ctDNA is informative as it can provide details on specific mutant genotypes similar to a traditional biopsy but in a relatively less invasive way [16]. More importantly, it is also correlative to the total tumor burden [17], which can be used quantitatively to measure drug efficacy. The primary objective is to establish the predictive value of ctDNA in monitoring patients on afatinib who have progressed on currently available EGFR TKIs gefitinib or erlotinib. Our results clearly showed that variations in ctDNA quantities are directly related to drug response in our patient cohort. This lays the foundation for using ctDNA for clinical monitoring as well as treatment intervention.
Methods and Materials
Study Design and patient characteristics
All patients provided written informed consent to participate in the study. The institutional review board approved all procedures for patient recruitment and sample extraction. This is a single-arm trial to determine the potential usefulness of ctDNA as a biomarker for patients after initial treatment with 1st generation EGFR-TKIs (erlotinib and gefitinib). The primary end point is overall survival (OS). Patients enrolled in the trial had either one or more of the activating EGFR mutations (L858R or exon 19 deletion) as determined by the molecular profiling of primary tissue samples. Patients with de novo T790M mutations are excluded from the study. Peripheral blood of 3 mL was extracted at specific time points from each patient for ctDNA analysis during the afatinib treatment process.
Treatment
Patients received a single oral starting dose of afatinib at 40 mg daily after acquiring resistance to 1st generation EGFR TKI erlotinib and/or gefitinib. Acquired resistance was defined using the Jackman’s clinical criteria [18], which is having previous treatment with either erlotinib or gefitinib; having positive EGFR mutations; having systemic progression of disease while on erlotinib or gefitinib for the last 30 days; and having no intervening systemic therapy. Treatment continued until progressive disease (PD) or no longer tolerated by patient due to adverse events. The drug is administered at least 1 hour before or 2 hours after a meal.
Blood sample processing and detection of mutant EGFR
ctDNA was recovered from peripheral blood of patients during patient follow up visits. 3 mL of blood was drawn in EDTA vacutainers and processed immediately. Briefly, blood samples was centrifuged at 2000 g for 10 minutes at 4 °C. Subsequently, the supernatant was removed and spun down again using the same conditions to remove any traces of cells in the plasma. Cell free DNA was extracted using the Qiagen’s QIAmp Circulating Nuclei Acid kit (Qiagen Inc., USA) following manufacturer’s instructions. The samples was stored at -20 °C before molecular profiling.
Validation runs on ddPCR of EGFR mutations
Detection of mutant EGFR was done using the QX200 droplet digital PCR (ddPCR) system (Bio-Rad Laboratories Inc., USA). Every sample was tested for L858R, exon 19 deletion and T790M. The procedures for processing and primers design were adopted from Oxnard et al [19]. Briefly, TaqMan PCR reactions were prepared from the 2× mastermix and custom TaqMan probes/primers made specific for each mutation in a 25 ul reaction. After droplet generation, samples were transferred to a PCR plate and amplified. After PCR, the plate was read on the droplet reader and data analyzed using the QuantaSoft software (Bio-Rad Laboratories Inc., USA). Standard curves of each mutation were prepared using L858R plasmids, PC-9 genomic DNA, T790M plasmids and wildtype EGFR plasmids. Spiked conditions varied in the range of 10 to 1 000 mutant copies, which simulates a low DNA template input. Addition validations were done with spiked mutant DNA of 100 copies in 1000 equivalent genome background. Each data point of the validation experiments was independently performed 5 times.
Statistical Analyses
The primary purpose of the study is to examine the relationship between plasma EGFR mutation status and the clinical effectiveness of afatinib treatment. Baseline concordance rate was calculated as the number of samples with matching results in both the primary tissue and initial plasma samples. The comparison analysis after administering afatinib to each patient was performed with the Student’s t test. OS calculations were done using the Kaplan-Meier methods and the hazard ratio between different patient groups were compared using the log-rank test. The categorical variables were presented as mean ± standard deviation unless otherwise defined. All statistical analysis was performed with the Prism Software (GraphPad, USA).
Results
Patient characteristics
A total of 200 patients with advanced NSCLC were enrolled in the study. The patient demographics and mutational profiles are provided in Table 1. EGFR mutational profiling was performed during routine clinical testing on tissue biopsies. The pathological specimens were retrieved using either fine needle aspiration or core needle biopsy. Only patients with positive EGFR mutations were selected for the study. Majority of patients had at least one positive activating EGFR mutation and the most prevalent were exon 19 deletion and L858R point mutation. Patients with de novo T790M mutations were excluded from the study.
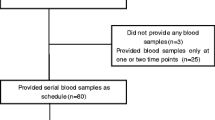
The baseline blood sample for each patient was extracted prior to treatment with EGFR-TKIs. Majority of the patients received gefitinib (70%), 13% had erlotinib and the rest received both. Figure 1A shows the flowchart of the study design and the blood sampling frequency. 168 patients (84%) had acquired TKI resistance according to Jackman’s clinical criteria [18] and most of them were subsequently monitored to detect ctDNA variations. The first blood sampling was performed before administering afatinib. Serial blood samplings were then taken whenever possible at regular intervals of 2 months during each patient follow up visits until treatment ceased. A total of 720 plasma samples were analyzed from the entire patient cohort of 200 individuals. A subset of 128 patients consented to be subsequently monitored for changes to the EGFR mutation profiles during afatinib treatment.

Blood sampling workflow and EFGR ctDNA characteristics prior to Afatinib treatment. a Study design and sample availability for ctDNA mutational analysis.b Baseline ctDNA measurements of the patient cohort. c Shift in molecular profiles showing a large proportion of T790M positive patients prior to afatinib initiation
Detection of ctDNA for EGFR mutations at baseline
The study utilized ddPCR to detect ctDNA within total cell free DNA as the assay has good sensitivity compared with other methods [20]. This is important as the expected quantity of ctDNA is low and accompanying wildtype DNA can be overwhelming [21]. In order to validate the assay, spiked samples of mutant DNA in increasing quantities were tested. The results showed good linearity of spiked input conditions for all EGFR mutations. Using linear regression, the r2 values for L858R, exon19 deletion and T790M were 0.985, 0.995 and 0.973 respectively (Supplementary Figure 1). This demonstrated excellent correlation and sensitivity to accurately detect the amounts of spiked input materials. Additionally, it was determined that in a 1000 equivalence genome background, 100 mutant copies can be reliably detected (Supplementary Fig. 2).
With the assay validated, we embarked on the molecular testing of clinical specimens. The baseline results compared the initial tissue molecular analysis of tumor biopsy with ctDNA from blood plasma. Overall concordance rate for EGFR mutations between the different types of samples was 93.5% as noted in Table 2. The result demonstrated the plasma samples were a good representation of the genotype. Each data point was deemed positive if the patients had the same L858R mutation or exon 19 deletion detected from tissue analysis. Individual concordance rate was 96.2% and 91.7% for L858R and exon 19 deletion respectively. Of the tissue and plasma analysis, there were 13 false negative results out of a total of 200 samples. Mutation specific concordance for T790M for both tissue and plasma samples was 100%.
Variations in ctDNA among patients at baseline are shown in Fig. 1b. We observed a fairly large spread of data points for the EGFR mutations. For the patient group harboring L858R, the concentration of mutant DNA ranges from 0 to more than 3000 copies/mL and average detected quantity among these 79 patients was 1422 ± 766 copies/mL. For exon 19 deletion, the detected concentration within the group of 121 patients ranges from 0 to more than 5000 copies/mL. The mean detected quantity was 2000 ± 1229 copies/mL. In addition, we observed good concordance of the tissue and blood plasma analysis for T790M profiles. All samples tested negative for T790M mutations using blood plasma, which was fully concordant with the tissue analysis.
Dynamic changes of EGFR mutation profile prior to afatinib treatment.
128 patients from the initial cohort who met the drug resistance criteria and consented to further participate in the study had their blood taken prior to afatinib treatment. We observed a substantial change in mutant DNA profile using plasma ctDNA within this patient cohort as depicted in Fig. 1c. The most striking observation was the emergence of T790M positivity among these patients. 47% of the study population was tested positive for T790M ctDNA at this sampling time point. The mutant ctDNA concentration among this subpopulation ranges from approximately 100 to more than 1 000 copies/mL and average detected amount was 660 ± 311 copies/mL as shown in Fig. 2a.

Quantitative measurements of ctDNA using ddPCR prior to afatinib treatment. a Molecular profile of patients with L858R, Exon 19 deletion and T790M before treatment. b Sub-analysis of ctDNA quantities between T790M positive and negative cohorts. T790M positive patients are likely to have higher concentration of corresponding L858R or Exon 19 deletion mutant DNA
Furthermore, we observed a statistically significant increase of mutant DNA concentrations detected within these participants (Fig. 2a). For the patient group that harbored L858R, the average increase was more than 30% and for exon 19 deletions, the surge was more than 40%. Interestingly, we noticed the variations were strongly associated with the presence of T790M mutation. As shown in Fig. 2b, the average increases were 54% and 89% for L859R and exon 19 deletion respectively for T790M positive patients.
Monitoring of response to afatinib shows clinical correlation
The first blood sampling for each patient was two to four weeks after starting treatment on afatinib. Thereafter, serial extraction of peripheral blood was done every 2 months interval. Among the patients that had their plasma results serially monitored, we observed a drop in mutant DNA concentration for 46% of the cohort immediately after treatment as shown in Fig. 3a. The L858R positive group had 38% of patients showing a decline while the exon 19 deletion group had 53%. The decline in ctDNA concentration was significant and ranges from 26 to 524 copies/mL. Of note, patients positively identified for T790M before the 2nd line treatment of afatinib had consistently detectable mutant signature. Mean mutant DNA concentration remained close to the reference analysis prior to afatinib and paired Student’s t test showed no significant difference within this group (Fig. 3b). No new cases of T790M positive patients emerged during the first plasma sampling after starting on afatinib but 3 more cases were found during subsequent serial monitoring.

Profiling of patients after afatinib treatment. a Measurement of L858R and Exon 19 deletion mutant DNA increment immediately (1st time point) after afatinib initiation. The data highlighted a significant number of patients having a decline in ctDNA levels after treatment. b Quantitative measure of ctDNA levels at different serial time points. c Quantification of T790M mutant DNA from T790M positive patients discovered prior to treatment
We tabulated the survival outcome for different groups of patients as the endpoint measure of this study. Median OS was 29.5 months for the entire cohort of 128 patients who underwent plasma monitoring as shown in Fig. 4a. We performed a series of subgroup analysis comparing patients with different profiles. Specifically, we analyzed the EGFR mutation type of patients who initially had L858R mutation with exon 19 deletions; the patient group who developed T790M resistance prior to afatinib; and patients who had registered a drop of 10% or more in ctDNA concentrations within the first 2 measured time points after administering afatinib.

Overall survival of patients receiving afatinib treatment in our study. a Collective results of all patients who were serially monitored using plasma ctDNA. b Sub-analysis of patient groups initially with activating EGFR mutations for L858R or Exon 19 deletions. c Sub-analysis of patient groups with T790M signature determined by ctDNA analysis prior to afatinib treatment. d Sub-analysis of patient groups with decrease in their ctDNA levels immediately after treatment show better survival outcome
The median OS for patients who were initially L858R positive was 29 months as compared with 30 months for patients who had exon 19 deletion at baseline. Hazard ratio was determined to be 1.06 (95% CI 0.69 to 1.63), with the p value 0.80, using a log rank test as depicted in Fig. 4b. For the patient group who were found to harbor T790M prior to 2nd line treatment, the median overall survival was 27 months, compared with 35 months for the cohort without the TKI resistance mutation. Hazard ratio was calculated to be 1.79 (95% CI 1.23 to 2.89) and the p value 0.005 (Fig. 4c). For the last group comparison of patients who saw a drop in ctDNA concentrations, the median OS was 34 months whereas the remaining patient group had median OS of 27 months. The calculated hazard ratio for this group was 1.59 (95% CI 1.029 to 2.456) with the p value 0.037 (Fig. 4d).
Discussion
The need for active clinical monitoring of cancer treatment to address the dynamic changes of EGFR mutations is crucial for measuring treatment efficacy and clinical intervention. Our study aims to show that ctDNA measurements is an effective means that correlates with 2nd line treatment using afatinib for NSCLC patients.
In our baseline concordance measurements of EGFR mutations via tissue biopsies and plasma DNA, we had demonstrated a significantly high concordance rate of 93.5% for L858R and deletions in exon 19. We had also achieved 100% concordance for baseline samples on T790M, a mutation that confers TKI resistance. This is highly suggestive that ctDNA is representative of the primary tumor and can be exploited for detection and monitoring as it is easily accessible via peripheral blood collection. Our results are also consistent with numerous other studies on NSCLC who saw a high concordance rate comparing plasma DNA with the matched tissue biopsy molecular profiling [22–25]. For instance, Kimura et al. demonstrated that plasma DNA achieved a 92.9% concordance rate with the primary tissue on a small sample size using the Scorpion Amplification Refractory Mutation System (ARMS) technology. Although the detection method is different, we have shown on a larger sample group that our concordance rate is similar. Multiple platforms have been demonstrated to reliably detect ctDNA from NSCLC [26], and our choice of using ddPCR is attributed to its low sample detection limit [19] that is crucial for detecting minute amounts of ctDNA [27]. We had determined in our validation trials on spiked sample controls that the assay readily detected mutant DNA of 100 copies within 1000 genome equivalent background. This demonstrated the excellent sensitivity of the assay to pick out low frequency mutations within each clinical specimen.
Our study further extends to understand how variations in ctDNA of the most commonly found EGFR mutations are affected by afatinib treatment. Afatinib has been revealed in a number of phase II trials to benefit NSCLC patients who acquired EGFR TKI resistance [15, 28–30]. We have demonstrated that serial monitoring of plasma samples can capture the dynamic changes during treatment that is crucial for clinical intervention. Of the 128 patients that were examined, we detected a significant number of individuals emerging with T790M positive secondary mutation. This highlighted a major change within the molecular profiles of these patients that was distinctly different from their baseline analysis. The initial absence of T790M from both the primary tissue and plasma cell free DNA suggested that the event was likely an acquired phenomena. Indeed, this is coherent with prior studies by Zhang et al [31] who showed that PC9 cells (lung adenocarcinoma cell line) acquired T790M after chronic exposures to increasing doses of gefitinib. In our cohort, we hypothesize the mechanism of the secondary resistance mutation (T790M) emergence is likely a result of the exposure to TKI therapy.
There were also fluctuations to the quantities of their initial EGFR mutations (Fig. 3b and Supplementary Figure 3), which can be representative of treatment efficacy. Specifically, we observed a strong correlation with T790M mutations, with patients who acquired the secondary EGFR mutation having the largest increase in mutant DNA levels. The results are highly suggestive of the reduced effectiveness of the 1st generation TKIs in managing the disease and the need for intervention such as switching to 2nd generation TKIs earlier.
Most patients who were subsequently placed on afatinib were given a dosage of 40mg daily. Patients who experienced adverse events/reactions had their dose reduced by 10mg. The most common adverse effects include diarrhea and skin rashes. Our parallel measurements of ctDNA during the treatment process yielded several interesting observations. 46% of the patient cohort registered a drop in ctDNA quantities and the corresponding OS measurements were favorable for these patients. Median OS was higher by 7 months as shown in Fig. 4d. Patients in this favorable group saw their ctDNA levels fall within 4 months after starting their 2nd line therapy. This is coherent with the LUX-Lung 4 study [15] that showed patients who responded on afatinib had a median progression free survival (PFS) of 4.4 months and responses by tumor imaging were seen within 8 weeks of afatinib initiation. In our study, our measured ctDNA quantities reduced by an average of 14% for the favorable patient group within 4 months (Supplementary Fig. 3). All patients within this favorable group registered a drop in ctDNA concentrations after afatinib initiation, albeit at differing amounts in the first plasma sampling cycle after afatinib treatment. For some patients, the declines in ctDNA levels were more pronounced at the 2nd plasma analysis. By the third sampling cycle, all patients had higher ctDNA counts than before afatinib treatment.
T790M remains the most crucial parameter that dictates treatment efficacy as shown in Fig. 4. Variations in T790M ctDNA quantity immediately after initiation of afatinib remained fairly stable (Fig. 3c) and could be consistently detected. Patients found to be T790M positive prior to afatinib treatment had median OS much lesser than the patient group without the secondary EGFR mutation. Similar to other studies [23, 32], we observed worse prognosis for the T790M positive patient group detected via ctDNA analysis. Maheswaran et al for instance observed that the presence of T790M prior to treatment using 1st generation TKI gefitinib and erlotinib was 7.7 months versus 16.5 months for TKI naïve patients [32]. Taken together, our results also suggest that T790M profiling before treatment can be a useful pretreatment marker. This is to classify patients who are not likely to achieve durable clinical response even to 2nd generation TKIs. In the current study, we advocate the use of ctDNA in the treatment monitoring of NSCLC patients undergoing 2nd line therapy for its relatively less invasive approach to tumor tissue extraction. Using ctDNA, multiple blood samplings throughout the treatment cycle can be performed to complement current monitoring regimes such as radiologic imaging.
One limitation of the current study is the lack of matched tissue biopsies at different blood sampling time points. A series of real time repeat biopsies would aid to confirm the dynamic changes that were observed in ctDNA analysis. However, this was technically difficult as the conditions of most of our patients at this stage were too weak and unsuitable for further repeat biopsy. Most patients were also reluctant for further surgical procedures. This concurs with the observations from other studies [33, 34] where getting access to repeat biopsy are challenging. ctDNA thus could complement this limitation in disease management by providing a less invasive procedure for disease profiling. Another interesting trial would be to vary the blood sampling frequency. This is to ascertain the optimal interval to capture significant events like the emergence of T790M that might lead to faster clinical intervention.
Our study nonetheless addressed a critical phase of NSCLC treatment using afatinib as the 2nd line therapy for patients who had acquired resistance to gefitinib or erlotinib. CtDNA is clearly an attractive means to actively probe the dynamics of the disease. This presents a complementary treatment analysis tool and can be applied to patients whose conditions do not allow for a biopsy.
Conclusion
In summary, our results clearly show that ctDNA is highly representative of the characteristics of key activating EGFR mutations at baseline, which confers their close connection to the primary tumor. The active monitoring of ctDNA profiles for each patient receiving therapy will aid to determine treatment efficacy and allow for quicker clinical intervention. In measuring the treatment efficiency of afatinib, the decrease in ctDNA levels was evidently linked to better OS outcome. Our analysis also showed that the patient group with the secondary EGFR T790M mutation prior to afatinib treatment had worse survival outcome. These results demonstrate that ctDNA is effective to guide TKI treatment for EGFR positive patients and future studies using this approach can be validated for a prospective clinical interventional study to determine the usefulness as a prognostic biomarker.
References
Gazdar AF (2009) Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 28(Suppl 1):S24–S31. doi:10.1038/onc.2009.198
Shigematsu H, Gazdar AF (2006) Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int J Cancer 118(2):257–262. doi:10.1002/ijc.21496
Shi Y, Au JS, Thongprasert S, Srinivasan S, Tsai CM, Khoa MT, Heeroma K, Itoh Y, Cornelio G, Yang PC (2014) A prospective, molecular epidemiology study of EGFR mutations in Asian patients with advanced non-small-cell lung cancer of adenocarcinoma histology (PIONEER). Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 9 (2):154-162. doi:10.1097/JTO.0000000000000033
Kris MG, Natale RB, Herbst RS, Lynch TJ Jr, Prager D, Belani CP, Schiller JH, Kelly K, Spiridonidis H, Sandler A, Albain KS, Cella D, Wolf MK, Averbuch SD, Ochs JJ, Kay AC (2003) Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA 290(16):2149–2158. doi:10.1001/jama.290.16.2149
Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S, Smylie M, Martins R, van Kooten M, Dediu M, Findlay B, Tu D, Johnston D, Bezjak A, Clark G, Santabarbara P, Seymour L, National Cancer Institute of Canada Clinical Trials G (2005) Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med 353(2):123–132. doi:10.1056/NEJMoa050753
Barker AJ, Gibson KH, Grundy W, Godfrey AA, Barlow JJ, Healy MP, Woodburn JR, Ashton SE, Curry BJ, Scarlett L, Henthorn L, Richards L (2001) Studies leading to the identification of ZD1839 (IRESSA): an orally active, selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to the treatment of cancer. Bioorg Med Chem Lett 11(14):1911–1914
Wakeling AE, Guy SP, Woodburn JR, Ashton SE, Curry BJ, Barker AJ, Gibson KH (2002) ZD1839 (Iressa): an orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res 62(20):5749–5754
Fukuoka M, Yano S, Giaccone G, Tamura T, Nakagawa K, Douillard JY, Nishiwaki Y, Vansteenkiste J, Kudoh S, Rischin D, Eek R, Horai T, Noda K, Takata I, Smit E, Averbuch S, Macleod A, Feyereislova A, Dong RP, Baselga J (2003) Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected]. J Clin Oncol Off J Am Soc Clin Oncol 21(12):2237–2246. doi:10.1200/JCO.2003.10.038
Perez-Soler R, Chachoua A, Hammond LA, Rowinsky EK, Huberman M, Karp D, Rigas J, Clark GM, Santabarbara P, Bonomi P (2004) Determinants of tumor response and survival with erlotinib in patients with non--small-cell lung cancer. J Clin Oncol Off J Am Soc Clin Oncol 22 (16):3238-3247. doi:10.1200/JCO.2004.11.057
Kosaka T, Yatabe Y, Endoh H, Yoshida K, Hida T, Tsuboi M, Tada H, Kuwano H, Mitsudomi T (2006) Analysis of Epidermal Growth Factor Receptor Gene Mutation in Patients with Non–Small Cell Lung Cancer and Acquired Resistance to Gefitinib. Clin Cancer Res 12(19):5764–5769
Balak MN, Gong Y, Riely GJ, Somwar R, Li AR, Zakowski MF, Chiang A, Yang G, Ouerfelli O, Kris MG, Ladanyi M, Miller VA, Pao W (2006) Novel D761Y and Common Secondary T790M Mutations in Epidermal Growth Factor Receptor–Mutant Lung Adenocarcinomas with Acquired Resistance to Kinase Inhibitors. Clin Cancer Res 12(21):6494–6501
Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, Padera RF, Shapiro GI, Baum A, Himmelsbach F, Rettig WJ, Meyerson M, Solca F, Greulich H, Wong, K K (2008) BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 27 (34):4702-4711. doi:http://www.nature.com/onc/journal/v27/n34/suppinfo/onc2008109s1.html
Solca F, Dahl G, Zoephel A, Bader G, Sanderson M, Klein C, Kraemer O, Himmelsbach F, Haaksma E, Adolf GR (2012) Target Binding Properties and Cellular Activity of Afatinib (BIBW 2992), an Irreversible ErbB Family Blocker. J Pharmacol Exp Ther 343(2):342–350
Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, Harris PL, Driscoll DR, Fidias P, Lynch TJ, Rabindran SK, McGinnis JP, Wissner A, Sharma SV, Isselbacher KJ, Settleman J, Haber DA (2005) Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A 102(21):7665–7670
Katakami N, Atagi S, Goto K, Hida T, Horai T, Inoue A, Ichinose Y, Koboyashi K, Takeda K, Kiura K, Nishio K, Seki Y, Ebisawa R, Shahidi M, Yamamoto N (2013) LUX-Lung 4: a phase ii trial of afatinib in patients with advanced non–small-cell lung cancer who progressed during prior treatment with erlotinib, gefitinib, or both. J Clin Oncol 31(27):3335–3341
Taniguchi K, Uchida J, Nishino K, Kumagai T, Okuyama T, Okami J, Higashiyama M, Kodama K, Imamura F, Kato K (2011) Quantitative Detection of EGFR Mutations in Circulating Tumor DNA Derived from Lung Adenocarcinomas. Clin Cancer Res 17(24):7808–7815. doi:10.1158/1078-0432.ccr-11-1712
Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, Kinzler KW, Vogelstein B, Diaz Jr LA (2008) Circulating mutant DNA to assess tumor dynamics. Nature Medicine 14 (9):985-990. http://www.nature.com/nm/journal/v14/n9/suppinfo/nm.1789_S1.html
Jackman D, Pao W, Riely GJ, Engelman JA, Kris MG, Janne PA, Lynch T, Johnson BE, Miller VA (2010) Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol Off J Am Soc Clin Oncol 28(2):357–360. doi:10.1200/JCO.2009.24.7049
Oxnard GR, Paweletz CP, Kuang Y, Mach SL, O'Connell A, Messineo MM, Luke JJ, Butaney M, Kirschmeier P, Jackman DM, Jänne PA (2014) Noninvasive Detection of Response and Resistance in EGFR-Mutant Lung Cancer Using Quantitative Next-Generation Genotyping of Cell-Free Plasma DNA. Clin Cancer Res 20(6):1698–1705
Watanabe M, Kawaguchi T, S-I I, Ando M, Tamiya A, Kubo A, Saka H, Takeo S, Adachi H, Tagawa T, Kakegawa S, Yamashita M, Kataoka K, Ichinose Y, Takeuchi Y, Sakamoto K, Matsumura A, Koh Y (2015) Ultra-Sensitive Detection of the Pretreatment EGFR T790M Mutation in Non–Small Cell Lung Cancer Patients with an EGFR-Activating Mutation Using Droplet Digital PCR. Clin Cancer Res 21(15):3552–3560
Kidess E, Heirich K, Wiggin M, Vysotskaia V, Visser BC, Marziali A, Wiedenmann B, Norton JA, Lee M, Jeffrey SS (2015) Mutation profiling of tumor DNA from plasma and tumor tissue of colorectal cancer patients with a novel, high-sensitivity multiplexed mutation detection platform. Oncotarget 6(4):2549
Szpechcinski A, Chorostowska-Wynimko J, Struniawski R, Kupis W, Rudzinski P, Langfort R, Puscinska E, Bielen P, Sliwinski P, Orlowski T (2015) Cell-free DNA levels in plasma of patients with non-small-cell lung cancer and inflammatory lung disease. Br J Cancer 113(3):476–483. doi:10.1038/bjc.2015.225
Zheng D, Ye X, Zhang MZ, Sun Y, Wang JY, Ni J, Zhang HP, Zhang L, Luo J, Zhang J, Tang L, Su B, Chen G, Zhu G, Gu Y, Xu JF (2016) Plasma EGFR T790M ctDNA status is associated with clinical outcome in advanced NSCLC patients with acquired EGFR-TKI resistance. Sci Report 6:20913. doi:10.1038/srep20913
Bai H, Mao L, Wang HS, Zhao J, Yang L, An TT, Wang X, Duan CJ, Wu NM, Guo ZQ, Liu YX, Liu HN, Wang YY, Wang J (2009) Epidermal growth factor receptor mutations in plasma dna samples predict tumor response in chinese patients with stages iiib to iv non–small-cell lung cancer. J Clin Oncol 27(16):2653–2659
Kimura H, Suminoe M, Kasahara K, Sone T, Araya T, Tamori S, Koizumi F, Nishio K, Miyamoto K, Fujimura M, Nakao S (2007) Evaluation of epidermal growth factor receptor mutation status in serum DNA as a predictor of response to gefitinib (IRESSA). Br J Cancer 97(6):778–784. doi:10.1038/sj.bjc.6603949
Thress KS, Brant R, Carr TH, Dearden S, Jenkins S, Brown H, Hammett T, Cantarini M, Barrett JC (2015) EGFR mutation detection in ctDNA from NSCLC patient plasma: A cross-platform comparison of leading technologies to support the clinical development of AZD9291. Lung Cancer 90(3):509–515. doi:10.1016/j.lungcan.2015.10.004
Yong E (2014) Cancer biomarkers: Written in blood. Nature 511(7511):524–526. doi:10.1038/511524a
De Grève J, Moran T, Graas M-P, Galdermans D, Vuylsteke P, Canon J-L, Schallier D, Decoster L, Teugels E, Massey D, Chand VK, Vansteenkiste J (2015) Phase II study of afatinib, an irreversible ErbB family blocker, in demographically and genotypically defined lung adenocarcinoma. Lung Cancer 88(1):63–69. doi:10.1016/j.lungcan.2015.01.013
Bar J, Botser D, Navon R, Peled N, Biran H, Gal-Yam EN, Ben-Arieh S, Raskin S, Onn A A Single-arm phase II study of afatinib treatment for advanced EGFR-mutant non-small cell lung cancer (mNSCLC) patients (pts) with EGFR-tyrosine kinase inhibitor (TKI) resistance. In: ASCO Annual Meeting Proceedings, 2015. vol 15_suppl. p e19031
Lee JY, Sun JM, Lim SH, Kim HS, Yoo KH, Jung KS, Song HN, Ku BM, Koh J, Bae YH, Lee SH, Ahn JS, Park K, Ahn MJ (2016) A phase Ib/II study of afatinib in combination with nimotuzumab in non-small cell lung cancer patients with acquired resistance to gefitinib or erlotinib. Clin Cancer Res 22(9):2139–2145. doi:10.1158/1078-0432.CCR-15-1653
Zhang S, Zheng X, Huang H, Wu K, Wang B, Chen X, Ma S (2015) Afatinib increases sensitivity to radiation in non-small cell lung cancer cells with acquired EGFR T790M mutation. Oncotarget 6(8):5832
Maheswaran S, Sequist LV, Nagrath S, Ulkus L, Brannigan B, Collura CV, Inserra E, Diederichs S, Iafrate AJ, Bell DW, Digumarthy S, Muzikansky A, Irimia D, Settleman J, Tompkins RG, Lynch TJ, Toner M, Haber DA (2008) Detection of mutations in EGFR in circulating lung-cancer cells. N Engl J Med 359(4):366–377. doi:10.1056/NEJMoa0800668
Kim ES, Herbst RS, Wistuba II, Lee JJ, Blumenschein GR Jr, Tsao A, Stewart DJ, Hicks ME, Erasmus J Jr, Gupta S, Alden CM, Liu S, Tang X, Khuri FR, Tran HT, Johnson BE, Heymach JV, Mao L, Fossella F, Kies MS, Papadimitrakopoulou V, Davis SE, Lippman SM, Hong WK (2011) The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov 1(1):44–53. doi:10.1158/2159-8274.CD-10-0010
Jekunen AP (2015) Role of rebiopsy in relapsed non-small cell lung cancer for directing oncology treatments. J Oncol 2015:11. doi:10.1155/2015/809835
Acknowledgement
This work was supported by a research grant provided by the Jingzhou Central Hospital.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
All other authors declare no conflict of interest.
Author’s Contribution
J.H and J.M designed the experiments. J.H, W.T and X.T performed the experiments and ctDNA analysis. J.H and J.M interpreted the data and wrote the manuscript.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed Consent
Informed consent was obtained from all individual participants included in the study.
Electronic Supplementary Materials
Supplementary Figure 1.
(DOC 797 kb)
Supplementary Figure 2.
(DOC 166 kb)
Supplementary Figure 3.
(DOC 468 kb)
Rights and permissions
About this article

Cite this article
He, J., Tan, W., Tang, X. et al. Variations in EGFR ctDNA Correlates to the Clinical Efficacy of Afatinib in Non Small Cell Lung Cancer with Acquired Resistance. Pathol. Oncol. Res. 23, 307–315 (2017). https://doi.org/10.1007/s12253-016-0097-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12253-016-0097-y