
Abstract
Pheochromocytomas (Pheo) and paragangliomas (PGL) are rare tumors, with heterogeneous genetic background. In up to 30 % of all, apparently sporadic Pheo/PGL cases germline mutations can be identified in one of the 15 genes representing genetic susceptibility for Pheo/PGL. Malignancy is rare but it frequently associates with SDHB mutations. Our aim was to determine the prevalence of germline SDHx, SDHAF2, MAX and TMEM127 mutations in Hungarian patients with apparently sporadic Pheo/PGLs. Mutation screening of the SDHx, SDHAF2, MAX and TMEM127 genes was performed in 82 Hungarian patients with apparently sporadic Pheo/PGL using PCR and bidirectional Sanger sequencing. Disease-causing germline mutations were identified in 11 patients, of which 4 SDHB and 2 TMEM127 mutations were novel. Earlier development of Pheo/PGL, more malignant phenotype and multiple tumors were observed in genetically positive cases especially in those with SDHB mutations. The presence of bilateral or multiple tumors was the most predictive for identification of a pathogenic mutation. Together with cases harboring germline RET, VHL and NF1 mutations, Hungarian patients with Pheo/PGL exhibit a heterogeneous mutation spectrum, indicating that all of the Pheo/PGL susceptibility genes should be tested. Novel genotype-phenotype associations revealed by our study may contribute to improvement of diagnostic approaches and may help to achieve a better clinical follow up for patients with Pheo/PGL.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pheochromocytomas (Pheos) and paragangliomas (PGLs) are rare tumors; their incidence in the general population is approximately 2–8 cases/1 million/year. These tumors are arising from neural crest derived chromaffin cells, producing and secreting catecholamines. Pheo refers to tumors arising from the adrenal medulla, while PGLs originate from the sympathetic (e.g. organ of Zuckerkandl) or parasympathetic (e.g. carotid body) paraganglia [1].
Most of these tumors are sporadic, but 20–30 % of all cases are part of well characterized hereditary tumor syndromes [1, 2]. These syndromes include Multiple Endocrine Neoplasia 2 (MEN2), von Hippel-Lindau Disease (VHL), Neurofibromatosis type 1(NF1) and familial paraganglioma syndrome types 1–5 (PGL) caused by mutations of RET protooncogene (MEN2), VHL (VHL syndrome), neurofibromin (NF1) and genes encoding the subunits of succinate dehydrogenase enzymes (SDHD-PGL1, SDHC-PGL3, SDHB-PGL4 and SDHA-PGL5) [3–7]. Recently the list of genes involved in the genetic susceptibility of Pheo/PGL extended by identification of germline mutations in genes encoding kinesin family member 1B (KIF1B) [8], EGL nine homolog 1 (EGLN1) [9], the transmembrane protein 127 (TMEM127) [10, 11], MYC-associated factor X (MAX) [12], fumarate hydratase (FH) [13] and malate dehydrogenase type 2 (MDH2) [14] making Pheo/PGL a multigenic disorder.
Due to the large number of genes responsible for the development of Pheo/PGL the genetic testing remains a diagnostic challenge. Both laboratory work load and cost of testing of all these genes are still significant despite of the lower price of molecular biological reagents. Phenotype oriented guidelines allow us some priorization in the order of genes tested but after a negative result the remaining genes should be also examined. Therefore, it would be ideal that after exclusion of some genes based on the obvious phenotype features (i.e, because of typical manifestation the NF1 gene is rarely tested) all of the remaining genes would be tested at the same time. Recent technical improvements in sequencing technology - next generation sequencing (NGS) platforms - allow us to use whole exome or targeted resequencing of all these genes [15]. The usefulness of NGS has been demonstrated not only in resequencing of already known genes, but also in discoveries of novel genes associated with Pheo/PGL [12, 14, 16–19]. However, after an NGS-based analysis Sanger sequencing is used for confirmation of results and a negative NGS result does not exclude the possibility of mutations. Therefore, the “gold standard” methodology for identification of pathogenic mutation is the PCR amplification of coding region of target genes followed by Sanger sequencing. For large deletion analysis multiple ligation probe amplication (MLPA) should also be performed. In addition, the Endocrine Society clinical practice guideline recommend the use of a clinical feature-driven diagnostic algorithm to establish the priorities for specific genetic testing in Pheo/PGL patients with suspected germline mutations delivered within the framework of health care [20, 21].
Our centre oversees the majority of the Hungarian Pheo/PGL population and the genetic testing using routine molecular biological methods including Sanger sequencing of RET, VHL, SDHB, SDHC, SDHD, SDHAF2, MAX and TMEM127, and multiplex probe amplification (MLPA) for VHL and SDHx are performed in our laboratory. We present this current study in order to summarize the prevalence of germline mutations among Hungarian patients with apparently sporadic Pheos/PGLs and to evaluate the genotype-phenotype associations in our patients with novel germline SDHB, SDHC and TMEM127 mutations.
Materials and Methods
Patients
Our database containing the clinical and laboratory data of 129 patients diagnosed and followed up at the 2nd Department of Internal Medicine, Semmelweis University with clinical diagnosis of Pheo/PGL between 1998 and 2014 was reviewed in order to select cases for comprehensive genetic testing. Of these patients the clinical diagnosis was confirmed by pathological examination of the surgically removed tumor tissues in 92 cases. Mutation screening of the RET and VHL genes identified 4 RET mutation carriers and 4 patients with germline VHL mutations [22–24]. In two cases the specific phenotype features indicated neurofibromatosis type 1. These patients were excluded from this current analysis and SDHB, SDHC, SDHD, SDHAF2, MAX and TMEM127 mutation analysis was performed in 82 cases. The main demographic and pathological data are summarized in Table 1.
Genetic Testing of the RET, VHL, SDHB, SDHC, SDHD, SDHAF2, MAX and TMEM127 Genes Using Sanger Sequencing
After genetic counseling and obtaining informed consent all 82 patients underwent genetic testing for the SDHB, SDHC, SDHD, SDHAF2, MAX and TMEM127 using conventional methods including PCR followed by Sanger sequencing (22–24). Blood DNA was extracted using commercially available DNA extraction kits (DNS isolation from mammalian blood, Roche, or DNA isolation kit from blood, Qiagen LTD). Bidirectional DNA sequencing of all these genes and large deletion analysis of the SDHB, SDHC, and SDHD genes were performed using multiplex ligation probe amplification [24].
Results
Prevalence of Germline Mutations in Pheo/PGL Susceptibility Genes
Eleven patients were identified to carry mutation in one of the Pheo/PGL associated genes. Together with our previous data demonstrating mutations in RET (n = 4) and VHL (n = 4) genes, the prevalence of germline disease-causing mutations in Hungarian patients with apparently sporadic, non-syndromic Pheo/PGL is 21.1 % (Table 1). To detect pathogenic mutation, bilateral involvement and multiple tumors had the most predictive value. The prevalence of bilateral tumors was significantly higher in mutation carriers than in genetically negative cases (8 of 11, 72.8 % vs. 3 of 71, 2.1 %; p < 0.001).
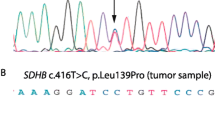
The mutation spectrum observed in our patients was heterogeneous, the most frequent mutations were detected in the SDHB gene (7 different of which 4 were novel mutations), Three patients had TMEM127 mutations (two novel) and one had mutation in the SDHD gene (Table 2). The chromatograms of all novel mutations identified are presented in Fig. 1. All novel SDHB mutation have been submitted to TCA Mutation Database and the new TMEM127 mutations to dbSNP database (http://chromium.lovd.nl/LOVD2/SDH/variants.php?select_db=SDHB&action= view&view = 0,000,838, http://chromium.lovd.nl/LOVD2/SDH/variants.php?select_db=SDHB& action = view&view = 0,000,839; http://chromium.lovd.nl/LOVD2/SDH/variants.php?select_ db = SDHB&action = view&view = 0,000,840;http://chromium.lovd.nl/LOVD2/SDH/ variants.php? select_db = SDHB& action = view&view = 0,000,841) and the novel.

Results of Sanger sequencing and chromatograms of novel germline variants identified in 6 patients with Pheo/PGL
No mutations in SDHC, SDHAF2, and MAX was identified in our patients.
Genotype-Phenotype Associations
Comparison of the main demographic and clinical data of the genetically positive and negative cases indicated that genetically positive patients were younger, their Pheo/PGL was more frequently malignant, and 72 % of cases had bilateral or multiple tumors (Table 1). As expected the malignancy was the highest (3 out of 7 cases) in patients with SDHB mutations. Two patients with mutations SDHB:c758G > A -Cys253Tyr- and the novel SDHB: c.586 T > G -Cys196Gly- were lost because of metastatic disease by the age of 35 years. In these patients multiple metastases in bone and liver were observed. In the third case with malignant PGL the novel SDHB: c728G > A Cys243Tyr mutation was identified. In this patient an intraabdominal PGL with multiple bone metastasis was diagnosed.
Another important finding was that the SDHB associated tumors were mainly intraabdominal PGLs (6 out of the 7 cases). In one case with the novel SDHB c607G > T Gly203Stop mutation pheochromocytoma and renal cell carcinoma with oncocytic feature was detected at age of 19 years. The solid architecture, cytoplasmic inclusions of flocculent material and intratumoral mast cells as the main characteristics for SDHB associated renal cell carcinomas could be identified (Fig. 2).

Immunohistochemical labeling of tumours associated with novel SDHB mutations. Both PGLs and renal cell carcinoma with oncocytic feature associated with SDHB mutations showed no SDHB immunohistochemical staining. Panel A. Intraabdominal PGL associated with SDHB: c.586 T > G (Cys196Gly), positive control: adrenaocortical cells; Panel B: paraganglioma associated with the SDHB: c.728G > A (Cys243Tyr) mutation, positive control: endothelial cells; Panel C: Renal cell cancer associated with the SDHB: c.607G > T (Gly203Stop) mutation. Entrapped non-neoplastic renal tubules showed positive immunohistochemical labeling for SDHB
Head-neck PGLs were detected in a patient harboring the SDHB: c286 + 1G/A mutation, and in a patient with SDHD c.147–148 insA frameshift mutation. In the later case an intraabdominal PGL was also removed. After 4–8 years follow-up no malignancy was observed in these cases.
TMEM127 mutations were detected in three patients. Two of them had Pheo (one bilateral) while in the third patient with the novel mutation (TMEM127: c467T > A, − Leu155Stop) Pheo and PGL of the head-neck region was also observed. These tumors showed no malignancy. The youngest patient harboring TMEM127 associated tumor was 22 years old.
Discussion
Pheo/PGLs are rare chatecholamine producing tumors. To date 15 genes have been implicated in genetic susceptibility, which are responsible for the 25–30 % of all Pheo/PGL cases. The American Society of Clinical Oncology have suggested that for patients with a ≥ 10 % chance for carrying a germline mutation genetic testing should be offered [20, 25]. Patients with Pheo/PGL are in this group. Currently the genetic analysis of patients with Pheo/PGL includes molecular genetic analysis of RET, VHL, SDHx, MAX and TMEM127 genes. MAX and TMEM127 were identified in 2010, and to date only few studies have been published about the prevalence of mutations in apparently sporadic cases [2, 10, 18, 26, 27]. Our current study was initiated to comprehensively analyze the prevalence of germline mutation in our cohort of histologically confirmed non-syndromic patients. Using conventional molecular biological methods we identified 11 germline mutation carriers, including six novel mutations. These results, together with our previous data on RET (n = 4) and VHL mutations (n = 4) in Hungarian patients with apparently sporadic, non-syndromic Pheo/PGL shows that 21.1 % of our patients carry mutation in one of the Pheo/PGL susceptibility genes [22, 24]. This finding is in line with previously reported data in other populations [2, 26] and with a recent review by Brito et al. [27].
The mutation spectrum observed in our cohort suggests that no founder mutation is present in the Hungarian population. Genetic studies performed in the past did not include the mutation testing of KIF1B, EGLN1, TMEM127, MAX or the recently identified MDH2 and their prevalence in apparently sporadic Pheo/PGL cases are lacking. Therefore, our study is also important from this aspect and the results demonstrated that in a population with heterogeneous genetic background the genetic screening should be performed for all of these genes.
The novel mutations identified in our cases are considered as disease-causing mutations, because they are either protein truncating mutations (TMEM127. c572delC, SDHB Gly203Stop and TMEM127, Leu155Stop) or they affect residues which are important for protein function and in the same codon other mutations have already been reported as pathogenic (SDHB p.Cys196Gly and p.Cys243Tyr) according to the TCA Cycle Gene Mutation Database (http://chromium.liacs.nl). These novel mutation are not listed in any database including dbSNP database (http://www.ncbi.nih.gov/SNP), exome variant server (http://evs.gs. washington.edu/EVS/, version v.0.0.30) and Exac variant (exac.broadinstitute.org/) databases. In addition a negative SDHB immunostaining of tumors associated with SDHB p.Cys196Gly, p.Cys243Tyr and Gly203Stop (Fig. 2) further supports the pathogenic role of SDHB mutations in these patients.
Genotype-phenotype associations confirmed that the malignant potential is frequently associates with SDHB mutations. The presentation and the course of the disease of our case with the SDHB Cys196Gly mutation were unique. In this case malignant PGL presenting as a primary PGL in the occipital bone was found. By reviewing the literature only one similar case was found. Kanai et. presented a 61-year-old male patient diagnosed with multiple paragangliomas including intracranial PGL and osteolytic lesion in the occipital bone. Despite surgical interventions and chemotherapy, the patient died in the fourth year after the diagnosis. No data about the genetic background of this case was reported but the similar behavior observed in these two cases may raise the pathogenic role of SDHB [28].
In addition, a more complex phenotype, including a rare concomitant tumor (Pheo/PGL and renal cell carcinoma) was found in another patient with SDHB mutation. Renal cell carcinoma with oncocytic feature has been reported as a hallmark of the SDHB associated renal cell carcinomas [21, 29]. In our patient the lack of SDHB staining confirmed the loss of SDHB protein in tumor tissue while it was kept in renal tubular cells. Based on our and Williamson’s results genetic testing of the SDHB gene should be offered for patients presenting with renal cell carcinoma with oncocytic features [21, 29].
The lack of mutation of SDHC gene is not entirely unexpected because our patient group consisted of patients having mostly intraabdominal PGLs and Pheos whereas SDHC mutations have been identified exclusively in head and neck PGLs [2, 18, 20]. In addition, sporadic head and neck PGLs may present with less symptoms and may possibly be underdiagnosed.
Mutations in SDHAF2, MAX and TMEM127 genes have been reported only in a very few cases [12, 27]. In our study no SDHAF2 and MAX mutations were found but TMEM127 mutations were detected in 3 patients of which 2 mutations proved to be novel. It seems particularly important that TMEM127 mutations were previously reported only in patients with adrenal Pheos, but in one of our patient having a novel TMEM127 mutation bilateral adrenal Pheos as well as glomus caroticum PGL were diagnosed. This new phenotype indicates that mutations of TMEM127 can also associate with head and neck PGLs [10, 11, 30]. In our study the two novel TMEM127 mutations were truncating mutations strongly suggesting their deleterious nature. The third TMEM127 mutation was detected in a 22-years-old) female patient presenting with unilateral adrenal Pheo. This mutation was already reported by Yao et al. and, surprisingly this seems to be the only TMEM127 mutation associated with malignant phenotype [11, 30]. Toledo et al. reported a six generation family with TMEM127 mutation and suggested that clinical surveillance in TMEM127 carriers should be started at age of 22 years. Our finding indicate that clinical surveillance should be started at earlier age.
In our mutation negative patients only three cases presenting with bilateral or multiple tumors had no disease causing mutation. These patients, together with the 12 patients with malignant phenotype (5 PGL and 7 Pheo) may have mutations of genes which were not investigated in the present study. Testing the KIF1B, EGLN1, FH, IDH2 and MDH2 genes by classical methods represents a significant work load and cost, therefore, next generation sequencing based methods would be desired.
The clinical follow-up of patients identified with pathogenic, germline mutation and their first-degree relatives is challenging. First of all, in the affected families for the first degree relatives genetic counseling followed by genetic testing should be offered. These tumours syndromes are inherited in an autosomal dominant manner, therefore the chance of inheriting the pathogenic variant is 50 %. The SDHD gene is maternally imprinted therefore the pathogenic variant is inherited from the paternal side, hence in children inheriting mutation from their mother the development of the disease is extremely unexpected. The penetrance of Pheo/PGL varies significantly between these syndromes. It seems to be very low for SDHA, SDHB, SDHC, SDHD and TMEM127 mutations but it is higher for RET, VHL and NF1 alterations. Of course the typical manifestations associating with RET, VHL and NF1 mutations are highly penetrant and several times preceed the development of Pheo (ie. mdullary thyroid cancer in RET mutation carriers, renal cell cancer, hemangioblastoma and retina angiomatosis in VHL carriers and skin lesions in NF1 mutation carriers). In these families the routine clinical follow-up includes regular checking for manifestation using laboratory and imaging techniques (summarized by Lenders, 20).
In summary, our current study presents results of a comprehensive mutational screening of a large series of patients with Pheo/PGL. The heterogeneous genetic background with six novel mutations observed in Hungarian patients is similar to other populations where no founder mutations are present. The genetic screening offered for Pheo/PGL patients in these populations should cover all of the genes identified to date but the first gene for testing should be the SDHB in cases with intraabdominal PGL especially with malignant phenotype. The novel genotype-phenotype associations revealed by our study may contribute to improvement of diagnostic approaches and may help to achieve a better clinical follow up of patients with Pheo/PGL.
References
Toledo RA, Dahia PL (2015) Next-generation sequencing for the diagnosis of hereditary pheochromocytoma and paraganglioma syndromes. Curr Opin Endocrinol Diabetes Obes 22(3):169–179
Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K, Januszewicz A, Eng C, Smith WM, Munk R, Manz T, Glaesker S, Apel TW, Treier M, Reineke M, Walz MK, Hoang-Vu C, Brauckhoff M, Klein-Franke A, Klose P, Schmidt H, Maier-Woelfle M, Peçzkowska M, Szmigielski C, Eng C (2002) Freiburg-Warsaw-Columbus Pheochromocytoma Study Group germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 346:1459–1466
Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, Richard CW 3rd, Cornelisse CJ, Devilee P, Devlin B (2000) Mutations in SDHD, a Mitochondrial Complex II Gene, in Hereditary Paraganglioma Science 287:848
Niemann S, Müller U (2000) Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet 26(3):268–270
Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, George E, Sköldberg F, Husebye ES, Eng C, Maher ER (2001) Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet 69(1):49–54
Hao HX, Khalimonchuk O, Schraders M, Dephoure N, Bayley JP, Kunst H, Devilee P, Cremers CW, Schiffman JD, Bentz BG, Gygi SP, Winge DR, Kremer H, Rutter J (2009) SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 28;:1139–42
Burnichon N, Brière JJ, Libé R, Vescovo L, Rivière J, Tissier F, Jouanno E, Jeunemaitre X, Bénit P, Tzagoloff A, Rustin P, Bertherat J, Favier J, Gimenez-roqueplo AP (2010) SDHA Is a Tumor Suppressor Gene Causing Paraganglioma. Hum Mol Genet 19:3011–3020
Yeh IT, Lenci RE, Qin Y, Buddavarapu K, Ligon AH, Leteurtre E, Do Cao C, Cardot-Bauters C, Pigny P, Dahia PL (2008) A germline mutation of the KIF1B beta gene on 1p36 in a family with neural and nonneural tumors. Hum Genet 124(3):279–285
Ladroue C, Carcenac R, Leporrier M, Gad S, Le Hello C, Galateau-Salle F, Feunteun J, Pouysségur J, Richard S, Gardie B (2008) PHD2 mutation and congenital erythrocytosis with paraganglioma. N Engl J Med 359(25):2685–2692
Qin Y, Yao L, King EE, Buddavarapu K, Lenci RE, Chocron ES, Lechleiter JD, Sass M, Aronin N, Schiavi F, Boaretto F, Opocher G, Toledo RA, Toledo SP, Stiles C, Aguiar RC, Dahia PL (2010) Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet 42(3):229–233
Yao L, Schiavi F, Cascon A, Qin Y, Inglada-Pérez L, King EE, Toledo RA, Ercolino T, Rapizzi E, Ricketts CJ, Mori L, Giacchè M, Mendola A, Taschin E, Boaretto F, Loli P, Iacobone M, Rossi GP, Biondi B, Lima-Junior JV, Kater CE, Bex M, Vikkula M, Grossman AB, Gruber SB, Barontini M, Persu A, Castellano M, Toledo SP, Maher ER, Mannelli M, Opocher G, Robledo M, Dahia PL (2010) Spectrum and prevalence of FP/TMEM127 gene mutations in pheochromocytomas and paragangliomas. JAMA 304:2611–2619
Comino-Méndez I, Gracia-Aznárez FJ, Schiavi F, Landa I, Leandro-García LJ, Letón R, Honrado E, Ramos-Medina R, Caronia D, Pita G, Gómez-Graña A, de Cubas AA, Inglada-Pérez L, Maliszewska A, Taschin E, Bobisse S, Pica G, Loli P, Hernández-Lavado R, Díaz JA, Gómez-Morales M, González-Neira A, Roncador G, Rodríguez-Antona C, Benítez J, Mannelli M, Opocher G, Robledo M, Cascón A (2011) Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet 43(7):663–667
Castro-Vega LJ, Buffet A, De Cubas AA, Cascón A, Menara M, Khalifa E, Amar L, Azriel S, Bourdeau I, Chabre O, Currás-Freixes M, Franco-Vidal V, Guillaud-Bataille M, Simian C, Morin A, Letón R, Gómez-Graña A, Pollard PJ, Rustin P, Robledo M, Favier J, Gimenez-Roqueplo AP (2014) Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum Mol Genet 23(9):2440–2446
Cascón A, Comino-Méndez I, Currás-Freixes M, Inglada-Pérez L, de Cubas AA, Currás-Freixes M, Tysoe C, Izatt L, Letón R, Gómez-Graña Á, Mancikova V, Apellániz-Ruiz M, Mannelli M, Schiavi F, Favier J, Gimenez-Roqueplo AP, Timmers HJ, Roncador G, Garcia JF, Rodríguez-Antona C, Robledo M, Cascón A (2015) Whole-exome sequencing identifies MDH2 as a new familial paraganglioma gene. J Natl Cancer Inst 11:107(5). doi:10.1093/jnci/djv053
Cao M, Sun F, Huang X, Dai J, Cui B, Ning G (2013) Analysis of the inheritance pattern of a Chinese family with phaeochromocytomas through whole exome sequencing. Gene 526(2):164–169
McInerney-Leo AM, Marshall MS, Benn DE, McFarlane J, Robinson BG, Brown MA, Leo PJ, Clifton-Bligh RJ, Duncan EL (2014) Whole exome sequencing is an efficient and sensitive method for detection of germline mutations in patients with phaeochromcytomas and paragangliomas. Clin Endocrinol 80(1):25–33
Crona J, Verdugo AD, Granberg D, Welin S, Stålberg P, Hellman P, Björklund P (2013) Next-generation sequencing in the clinical genetic screening of patients with pheochromocytoma and paraganglioma. Endocr Connect 2(2):104–111
Welander J, Andreasson A, Juhlin CC, Wiseman RW, Bäckdahl M, Höög A, Larsson C, Gimm O, Söderkvist P (2014) Rare germline mutations identified by targeted next-generation sequencing of susceptibility genes in pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 99(7):E1352–E1360
Fishbein L, Khare S, Wubbenhorst B, DeSloover D, D'Andrea K, Merrill S, Cho NW, Greenberg RA, Else T, Montone K, LiVolsi V, Fraker D, Daber R, Cohen DL, Nathanson KL (2015) Whole-exome sequencing identifies somatic ATRX mutations in pheochromocytomas and paragangliomas. Nat Commun 6:6140
Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, Naruse M, Pacak K, Young WF Jr; Endocrine Society (2014) Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 99(6):1915–1942
Ricketts C, Woodward ER, Killick P, Morris MR, Astuti D, Latif F, Maher ER (2008) Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst 100(17):1260–1262
Patócs A, Karádi E, Tóth M, Varga I, Szücs N, Balogh K, Majnik J, Gláz E, Rácz K (2004) Clinical and biochemical features of sporadic and hereditary phaeochromocytomas: an analysis of 41 cases investigated in a single endocrine centre. Eur J Cancer Prev 13(5):403–409
Igaz P, Patócs A, Rácz K (2009) Uncommon MEN2A phenotype in a patient with a RET protooncogene exon 10, codon 611 mutation. Clin Endocrinol 71(2):304–305
Gergics P, Patocs A, Toth M, Igaz P, Szucs N, Liko I, Fazakas F, Szabo I, Kovacs B, Glaz E, Racz K (2009) Germline VHL gene mutations in Hungarian families with von Hippel-Lindau disease and patients with apparently sporadic unilateral pheochromocytomas. Eur J Endocrinol 161:495–502
Fishbein L, Merrill S, Fraker DL, Cohen DL, Nathanson KL (2013) Inherited mutations in pheochromocytoma and paraganglioma: why all patients should be offered genetic testing. Ann Surg Oncol 20(5):1444–1450
Pęczkowska M, Kowalska A, Sygut J, Waligórski D, Malinoc A, Janaszek-Sitkowska H, Prejbisz A, Januszewicz A, Neumann HP (2013) Testing new susceptibility genes in the cohort of apparently sporadic phaeochromocytoma/paraganglioma patients with clinical characteristics of hereditary syndromes. Clin Endocrinol 79(6):817–823
Brito JP, Asi N, Bancos I, Gionfriddo MR, Zeballos-Palacios CL, Leppin AL, Undavalli C, Wang Z, Domecq JP, Prustsky G, Elraiyah TA, Prokop LJ, Montori VM, Murad MH (2015) Testing for germline mutations in sporadic pheochromocytoma/paraganglioma: a systematic review. Clin Endocrinol 82(3):338–345
Kanai H, Yamashita N, Hashimoto N, Ogawa K, Suzuki S (2012) A case of malignant paraganglioma presenting with skull metastases. No Shinkei Geka 40(8):711–716
Williamson SR, Eble JN, Amin MB, Gupta NS, Smith SC, Sholl LM, Montironi R, Hirsch MS, Hornick JL (2015) Succinate dehydrogenase-deficient renal cell carcinoma: detailed characterization of 11 tumors defining a unique subtype of renal cell carcinoma. Mod Pathol 28:80–94
Toledo SP, Lourenço DM Jr, Sekiya T, Lucon AM, Baena ME, Castro CC, Bortolotto LA, Zerbini MC, Siqueira SA, Toledo RA, Dahia PL (2015) Penetrance and clinical features of pheochromocytoma in a six-generation family carrying a germline TMEM127 mutation. J Clin Endocrinol Metab 100(2):E308–E318
Acknowledgments
This study has been funded by Hungarian Scientific Research Grant (PD100648 to Attila Patocs), Technology Innovation Fund, National Developmental Agency (KTIA-AIK-2012-12-1-0010). Attila Patocs is a recipient of “Lendulet” grant from Hungarian Academy of Sciences.
Author Contributions
AP: study design, genetic counseling, mutation analysis, wrote the manuscript; IL: reviewed the manuscript, NL, HB: mutation analysis with Sanger sequencing; ZS: immunohistochemical analysis of the SDHB-associated renal cell carcinoma, NS, GT, PI, MT: diagnosis and clinical follow-up of patients with Pheo/PGL; KR: genetic counseling and clinical follow-up of patients with Pheo/PGL, reviewed the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors have no conflict of interest to report.
Informed Consent
All patient underwent genetic counseling and written informed consent was obtained before genetic analysis.
Disclosure Summary
None
Rights and permissions
About this article

Cite this article
Patócs, A., Lendvai, N.K., Butz, H. et al. Novel SDHB and TMEM127 Mutations in Patients with Pheochromocytoma/Paraganglioma Syndrome. Pathol. Oncol. Res. 22, 673–679 (2016). https://doi.org/10.1007/s12253-016-0050-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12253-016-0050-0