
Abstract
Transforming growth factor β (TGF-β) superfamily consists of numerous cytokins that regulate various cellular processes. TGF-β, the prototype of the family, signals through its cell surface serine/threonin kinase receptors and besides its role in cell differentiation, migration, adhesion etc. it is also able to induce epithelial-mesenchymal (EMT) transition via both Smad- pathway and MAPK- pathway. Among the different types of epithelial-mesenchymal transition, type II that is described to be associated with wound healing, tissue regeneration, organ fibrosis and is induced upon inflammatory stimuli. It can be triggered by secretion of growth factors such as TGF-β, EGF. Different endocytic routes are used for the internalization of TGF-β ligand and its receptors and these pathways can control the activity of downstream events. Internalization via clathrin-coated vesicles promotes the signaling while the caveola-mediated endocytosis plays important role in the termination of the events, although the steps of the latter event are less clear. The early endosome is considered a clue compartment in promoting the signaling. Recently published data suggest that the early endosome plays crucial role in the termination of the TGFβ signaling as well. It is not only maintain a special environment for the effective signaling but can direct the internalized cargos towards degradative pathways (multivesicular bodies, lysosomes).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Transforming growth factor β (TGF-β) superfamily consists of numerous groups of cytokins that regulate a diverse set of cellular processes. Besides the TGF-β isoforms (TGF-β1, TGF-β2 and TGF-β3), members of the family also include the bone morphogenetic proteins (BMP), inhibin, myostatin, Nodal, GDF, GDNF, MIS (Müllerian Inhibiting Substance), each with different roles in cell differentiation, apoptosis, cell migration, adhesion during embryogenesis and in adult tissues [1].
TGF-β has also dual role depending on the cell type and the environment. While it is able to suppress cell growth in epithelial and hemopoetic cells by inducing G1 arrest, it also initiates cell proliferation and differentiation in mesenchymal cells. These cellular processes regulate the morphological plasticity of a cell and result in phenotypic change that is known as epithelial-mesenchymal transition (EMT) [2].
Three subtypes of EMT can be distinguished with different functional consequences. Besides epithelial-mesenchymal transition during embryogenesis (type I) and tumorigenesis (type III), type II EMT is associated with wound healing, tissue regeneration and organ fibrosis. It has been demonstrated that during inflammation many cells can trigger type II EMT through secretion of growth factors such as TGF-β, EGF [3]. Most prominent among these cells are the macrophages and activated resident fibroblasts that accumulate at the site of injury and release these growth factors [4]. TGF-β was described to induce EMT via both Smad 2/3-dependent pathway and MAPK-dependent pathway.
The biochemistry of TGF-β signaling is in the focus of many articles and well characterized. It is less clear, however in which cellular/cytoplasmic compartments the molecules along the downstream pathway are accomodated and how their localization changes during the signaling. Another question of great interest is wether the different compartments can be involved in regulating the pathway and if so, wether they can promote or suppress the signaling? The universal role of TGF-β in the different types of EMTs is evident. The subcellular localization of the elements of different signaling pathways that might be the clue to define the adequate cellular response to TGF-β indicating the importance of compartmentalization.
The Signaling Events (Smad-Dependent Pathway)
TGF-β, the prototype of the family, signals through its cell surface serine/threonin kinase receptors. Functionally and structurally type I and type II TGF-β receptors (TβR-I, TβRII) can be distinguished. The type II receptor is considered a constitutively active kinase (activated by autophosphorylation) while type I receptor contains a special GS domain the phosphorylation of which leads to the activation of the receptor.
In the prototypic TGF-β pathway, ligand binds to type II receptor and induces the formation of a heterotetrameric receptor complex within which TβR-II transphosphorylates and activates the type I receptor and the activated TβR-I initiates the Smad signal transduction pathway [1, 5, 6].
The Smad proteins can be divided into three classes based on their structural and functional differences. The 1) receptor-regulated (R) - Smad proteins are Smad 2,3 that are the only substrates for type I receptor kinases and further members of this group are Smad 1,5,8, that are phosphorylated by the activated BMP receptors. After phosphorylation, thus activation, the R-Smads associate with 2) common mediator (Co)-Smad protein, Smad4. They form oligomeric complexes and are transported to the nucleus to regulate the transcription of target genes together with other nuclear cofactors. The members of third class of Smads act as negative regulators of the signaling pathway, the 3) inhibitory (I)-Smads, Smad6 and Smad7 proteins [6–8].
Recent findings have demonstrated that accessory proteins interact with type I, type II receptors and Smad proteins [7]. An example is SARA (Smad anchor for receptor activation) that facilitates the association of R-Smads with TGF-β receptor at the plasma membrane, though it is predominantly localized to phosphatidylinositol 3-phosphate (PtdIns3P) rich early endosomes [9, 10]. Furthermore, some data suggest that SARA can interact with cell surface TβRs and in this way protects the complex from degradation [11–14].
Ligand binding to its cell surface receptors means not only the beginning of the signaling events through Smads but also triggers internalization of both ligand and receptors [11–15]. The receptor internalization is required for the initiation of downstream signaling. There are two main endocytic pathways through which the TGF-β ligand-receptor complex can be internalized. One of them is the well-characterized clathrin-mediated endocytosis and a less clear pathway is the lipid/caveolae-mediated endocytosis. Both types of pathways are used for the internalization of TβRs. It is already clear that via different internalization routes cells can control the number of surface-receptors and this is crucial for regulating the signaling, receptor turnover, the magnitude and duration of the events [11].
Clathrin-Mediated Endocytosis Promotes the Signaling
Internalization of most cell surface receptors is mediated by short specific sequences in their cytoplasmic domain. Tyrosine-containing sequences and di-leucin-based motifs function as internalization signals for clathrin-dependent endocytosis. These sequences can directly bind to the endocytic machinery and play important role in cargo enrichment on the clathrin-coated pit as well as in vesicle formation [16, 17]. Such internalization signals have also been identified in TGFβ receptors. Both TβRI and TβRII appear to be rapidly internalized. After receptor-ligand internalization in clathrin coated vesicles, the complex is targeted into early endosome antigene-1 (EEA1) positive endosomes. It promotes the signal transduction by recruiting the FYVE domain-containing proteins (like SARA). The C-terminal phosphorylation of R-Smads occurs in endosomes leads to their dissociation from both SARA and receptor [10, 18, 19]. Then the phosphorylated R-Smads can bind to Smad4 [20] forming the oligomeric complex that can enter into the nucleus to regulate target genes in association with other coactivators and corepressors.
The shuttling of TGFβ-induced Smad complexes between the cytoplasm and the nucleus is strictly regulated. The R- Smad and Co- Smad proteins have conserved Mad- homology 1 (MH1) and MH2 domains connected by a linker domain, while the I-Smads are lack of a distinct MH1 domain. The R-Smads and Co-Smad have a nuclear localization sequence (NLS) in their Mad-homology 1 (MH1) domain while their MH2 domain contains nuclear export signal (NES) and nuclear pore signal (NPS) as well [21] (Fig. 1). Phosphorylated Smad3 was shown to interact with importin-β1 of the nuclear pore and enters into the nucleus in a GTPase dependent manner [22, 23].

Functional domains of Smad proteins. The R- Smad (Smad2/3) and Co- Smad (Smad4) proteins have conserved Mad- homology 1 (MH1) and MH2 domains connected by a linker domain, while the I-Smad (Smad7) is lack of a distinct MH1 domain. The MH1 domain contains DNA-binding site (except for Smad2) and nuclear localization signal (NLS) and mediates interactions with different transcription factors to stabilize the nuclear Smad complex. MH2 domain is higly conserved among all Smads and it is responsible for receptor interaction, the nucleocytoplasmic shuttling of Smad proteins (NPS, NES) and also mediates the formation of Smad oligomer complexes and the interaction of other proteins such as SARA. The linker region contains phosphorylation sites allowing crosstalks with other signaling pathways and binds ubiquitin ligases (Smurf proteins) via the PY motif
Early endosomes (EE), however, provide not only specialized environment for signaling events in the TGF-β pathway by recruiting the signaling molecules [18], but they are important cellular compartments where the internalized cargo proteins and receptors are sorted. The main factor in this process is the acidifying pH of endosomes that helps in the dissociation of receptor and ligand. After dissociation, TGF-β receptors can recycle back to the plasma membrane with the help of Rab11 positive recycling endosomes [24]. It is important to emphasise that although clathrin-mediated endocytosis of TβRs can enhance Smad-mediated TGFβ signaling, it is still debated wether this process is required for the signaling [18, 19]
Caveola-Mediated Endocytosis Turns off TGF-β Signaling
Another internalization route into the cell is via caveolin-1 positive vesicles and it is also known that TβRs are localized in lipid rafts of the plasma membrane. Caveolae are small plasma membrane invaginations that play important role in many cellular functions including signal transduction, cellular growth control, apoptotic cell death. The main protein components of caveolae are the scaffolding proteins termed caveolin-1,-2,-3 [25, 26]. Complex events lie behind the regulation of the internalization pathway through caveolae and the intermediate compartments are still less clear. Earlier data showed that caveolae internalize into the cell, and form so-called caveosomes that were supposed not to communicate with other cellular compartments. According to this idea caveosomes would represent a cellular compartment the content of which could avoid lysosomal degradation [27, 28]. Recent data, however, have shown that caveolin-positive vesicles can also associate with early endosomes [29, 30] and caveosomes are most likely modified late endosomes or lysosomes, thus they are part of the classical endocytic pathway [31]. According to this, caveolar endocytosis can also provide a possible way for sequestering receptors [15]. Several lines of evidence support the idea that receptor-ligand internalization via the (non-classical) caveolar pathway turns off the TGFβ signaling events by targeting the receptor-ligand complex to lysosomal and/or proteasomal degradation [10, 11, 15]. This receptor degradation plays an important role in controlling the amount of receptors on the plasma membrane. The possible pathways targeting the signaling molecules towards degradation are not entirely known and most of the papers avoid the detailed discussion of these routes. The inhibitory Smad (I-Smad), Smad7 is one of the main regulator in the degradative events. I-Smad inhibits TGFβ signaling through multiple mechanisms as a decoy substrate forming a stable complex with receptors to prevent recruitment of R-Smads [32, 33] and also disrupts the functional Smad-DNA complex formation [34]. Smad7 exerts its negative effects at the level of the plasma membrane by competing with R-Smads for the receptor and also by recruiting the E3 ubiquitin ligase Smurf1/2 proteins to the active TβRs [32, 35, 36] to promote receptor ubiquitination and degradation [21]. (Smad7 has a putative NLS in its N-terminal region and resides in the nucleus in non-stimulated cells. In response to TGFβ stimulus, Smad7 leaves the nucleus in complex with the ubiquitin ligases, Smurf1/2 [21, 35–37]). The interaction of Smad7 and Smurf proteins with activated TβRs targets the complex to lipid rafts/caveolae and in this way the caveola-mediated endocytosis could promote receptor turnover and the termination of signaling [10]. TGFβ receptors after receptor ubiquitination have been shown to be degraded by both lysosomal and proteasomal machineries [35, 38]. Though limited data are available on factors controlling the proteasomal degradation of TGFβ receptors. Recent data showed that a GPI-anchored protein, CD109 functions as a TGFβ co-receptor, associates with caveolin-1, promotes the caveolar localization of the TGFβ receptors and might regulate its proteasomal degradation [39]. However, it is not clear how the caveolar endocytic machinery can drive receptors to the proteasomal pathway.
Besides TβRs, the stability of Smad proteins and caveolin is also controlled by ubiquitination suggesting the regulatory role of different ubquitination signals. While poliubiquitination is a sign that directs the cargos to proteasomes, mono/multiubiquitination is a signal for the entry of proteins via the endocytic pathways. Thus, ubiquitination, indeed plays essential role both in signal transduction and also to determine the way of degradation towards proteasomes or towards multivescular body/late endosome formation [40, 41].
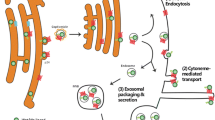
The lysosomal degradation of internalized cargos via caveolar endocytosis includes multivesicular body (MVB) formation and the early endosomes are the clue compartments of this process as well. Not only the receptor and ligand, but the caveolin itself can also be ubiquitinated, though the ubiquitinated caveolin is not directly degraded via multivesicular body (MVB) formation. Internalized cargo proteins are targeted first to early endosomes [31] indicating that MVB formation starts at the level of this cellular compartment. MVBs are formed when limiting membrane of endosomes invaginates and buds into the lumen of the organelles [42, 43]. A subset of membrane proteins within the limiting membrane of the endosomes are sorted into these invaginating vesicles and this sorting requires the inclusion of a 350 kDa complex, called ESCRT-1 (endosomal sorting complexes required for transport). The membrane of early endosome (EE) contains the ESCRT complex that recognizes and binds ubiquitinated cargos and initiates the transport of the cargos to late endosomes/multivesicular bodies. MVB sorting and the subsequent lysosomal degradation of cell surface receptors is therefore a critical mechanism for regulating the signaling events [44, 45]. Hence, the early endosome plays a central role not only promoting the TGF-β pathway, but it seems to be an important intermediate cellular compartment that helps to turn off the signaling as well (Fig. 2).

The early endosome (EE) plays central role in determinating the activity of TGF-β pathway. Internalization via clathrin-coated vesicles promotes the signaling as the EE provides a special environment where the phosphorylation of Smad 2/3 (which direct the downstream events) can occur. Afterwards the receptors can return back to the plasma membrane for reuse in Rab11 positive recycling endosomes. In contrast, the caveola-mediated endocytosis turns off the signaling events by associating with inhibitory Smad, Smad7 that recruits the E3 ubiquitin ligase Smurf 1/2. Thus, the ubiquitinated receptor-ligand complex is targeted for degradation. The early endosomes seem to be the intermediate organelles in this procedure as well. The early endosomal membrane contains the ESCRT-I complex necessary for multivesicular body (MVB) formation. Afterwards the multivesicular bodies fuse with lysosomes and the internalized cargos are degraded. Thus the early endosome plays crucial role not only in promoting but turning off the TGFβ signaling. The direct route of caveolae towards the proteasomes is still debated. CCV: clathrin-coated vesicle, CAV: caveola, P: phosphorylation, U: ubiquitination, MVB: multivesicular body, LE:late endosome
Negative Regulation Occurs at Different Cellular Levels
There are events both at the level of the cytoplasm and the nucleus that finally lead to the termination of signaling. During the degradative events (to turn off the signaling) various complex mechanisms occur in the cytoplasm. We have to consider that not only the receptor is ubiquitinated by Smurfs, but mono- and/or poliubiquitination can regulate the internalization and degradation rate of the caveolin-1 as well [31, 46].
Besides this, the level of Smad4 is also controlled by ubiquitination that regulates its stability and modulates its activity, thus the ubiquitin-mediated degradation both in proteasomes and lysosomes can regulate the stability of Smad proteins. Phosphorylation, dephosphorylation, acetylation, sumoylation can also have effect on the stability of Smads and can have effect on their proteasomal degradation [21]. It is well known that poliubiquitination targets the cargo to proteasomes. It is not clear whether the two independent pathways (MVB/lysosomal and proteasomal degradation) communicate with each other. Is there any cross talk between the two degradative routes? Another question is wether Smad7-Smurf proteins can be reused or is there any possibility for the recycling of these proteins during the caveolar internalization if we suppose that they also reach the early endosome? That might be another fine tuning for determinating the activity of the pathway.
In the nucleus an autoinhibitory feedback loop controls that parallel with the TGF-β stimulus, the transcription complex Smad2-Smad3/4 induces the expression of Smad7 and in this way it accumulates continuously in a concentration-dependent manner and helps to terminate the signaling [44]. Another mechanism at the nuclear level that blocks the transcription of target genes is the ubiquitination of the active transcription complex (Smad2–3/4) which targets them from the nucleus out to the cytoplasm for proteasomal degradation. This process is distinct from the Smurf-mediated ubiquitination in the cytoplasm. Further investigations will be required to describe the difference between nuclear and cytoplasmic degradation of Smad proteins and how they control the signaling [8].
There are data that indicate the role of estrogen-receptor (ER) α as a negative regulator of TGF-β pathway by increasing the degradation of nuclear Smad proteins. ERα forms a protein complex with Smad3/4 and ubiquitin ligases in the nucleus and enhances the degradation of the transcription complex by the ubiquitin-proteasome system [47].
TGF-β Induced Smad- Independent Pathways
Besides the (canonical) Smad- pathway, TGFβ activates other non-Smad signaling pathways such as Erk, JNK, p38 MAP kinase pathways in a cell-specific and context-dependent manner. MAPK pathways help and complete the process of TGFβ induced epithelial-mesenchymal transition, although the mechanism by which TGFβ activates these pathways and their biological consequences are poorly characterized [48, 49].
MAPK cascade is composed of several protein kinases that specifically phosphorylate and activate each other. The elements of the cascade are organized in levels that are termed MAP kinase kinase kinase (MAPKKK), MAP kinase kinase (MAPKK) and MAP kinase (MAPK). The activation of MAPK leads to its translocation to the nucleus where MAPK phosphorylates and activates its targets, e.x transcription factors. It is well known that MAPK pathways transmit extracellular signals to the nucleus to regulate different cellular processes [50–52]. However, it has recently been described that non-Smad signaling proteins (the elements of MAPK cascade) take part in the physiological responses of TGFβ as well by other different mechanisms: I) they can directly modify the activity of Smad proteins by e.x phosphorylation (p38 MAP kinase and JNK kinase have been reported to phosphorylate Smad2/3 and suppress their activity [53, 54]). II) They can directly interact or be phosphorylated by TβRs, hence a parallel signaling is initiated that might agonise Smad pathway or III) non-Smad proteins can directly be modulated by Smads that transmit signals to other pathways [49]. Emerging new data reflects the complexity of how the Smad- and non-Smad pathways are interconnected. The Erk MAPK phosphorylates the MH1 domain of Smad2 and blocks its nuclear translocation, thus transcriptional output. TGF-β induced JNK can also phosphorylate Smad3 and induces its translocation to the nucleus [55, 56]. The role of regulation of TβRs (phosphorylation, ubiquitination, sumoylation) is also necessary to be elucidated [57]. The phosphorylation of TβRII on tyrosine can contribute to the activation of TGFβ-induced p38 MAPK pathway and also the tyrosine phosphorylation of TβRI is necessary for the initiation of Erk MAPK pathway in response to TGFβ stimulus [58] (Fig. 3).

Summary of Smad-dependent and Smad-independent pathways that play role in TGF-β induced epithelial-mesenchymal transition. The formation of TGF-β ligand- receptor complex activates Smad2/3 proteins that form oligomer complexes with Smad4 and enter into the nucleus exerting their effects on target genes. Besides this signaling route, TGF-β ligand-receptor complex activates MAPK pathways (Erk, p38, JNK) that not only carry different extracellular signals towards the nucleus and contribute to the activity of transcription, but MAP kinases also modify the activity of Smad-proteins. The balance between activation of Smad proteins and MAPK pathways defines the cellular responses to TGF-β. For more details, see text.
Both Smad and MAPK signaling induced by TGF-β work together in a complex cellular network and the subcellular localization of both Smad and non-Smad proteins play an important role to define the final outcome for different extracellular stimuli. The signaling elements of MAPK pathways (MAPKKKs, MAPKKs,) are found at the plasma membrane and on endosomes, while the activated MAPKs are bounded to endosomal membranes. Thus, endosomes are crucial cytoplasmic compartments; as they create a platform and a special environment for the signaling molecules they can orchestrate the spatial and temporal regulation of different signaling routes [59, 60].
By now it is accepted that TGFβ induced Smad activation occurs in both lipid rafts/cavolae and non-lipid rafts, but a recent observation suggests that activation of MAPK in lipid rafts/cavolae is specially required for TGFβ induced EMT [61]. The role of raft compartments and endosomes is best charactarized in the Raf-MEK-Erk MAPK pathway. Raf kinases are localized near to the plasma membrane in the cytoplasm through interactions with different anchoring and scaffolding proteins or lipid compounds [62]. MEKs are localized in the cytoplasm of resting cells due to their nuclear export signal (NES). They shuttle between the cytoplasm and the nucleus constantly and they serve as cytoplasmic anchors for Erks. With the help of adaptor protein p18, MEKs are localized in the lipid rafts of late endosomes indicating the importance of endosomal compartments. Upon stimulation, Erks dissociate from MEK and through the formation of homodimers Erks enter into the nucleus by active transport mechanism, while as a monomer it can enter into the nucleus by passive diffusion. The nuclear export of Erks is mediated by a MEK dependent active transport mechanism due to their nuclear export signal [51, 59, 60, 63–65] (Fig. 4).

The subcellular localization of Erk MAP kinase pathway. Activated MAPKKK (H-Ras) is localized in lipid rafts/caveolae (CAV). MEK proteins are bounded to lipid compartments of late endosomes through adaptor proteins and MEKs serve as anchors for Erk. Upon stimulation, Erk dissociates from MEK and enters into the nucleus by passive transport and is replaced to the cytoplasm with the help of MEK proteins that has nuclear export signal (NES). For more details, see text. EE: early endosome, CAV: caveola, MVB: multivesicular body, LE:late endosome, P:phosphorylation
Conclusions
TGF-β exerts its effects in a cell-specific and context-dependent manner. There have been growing number of articles and evidences that reflected the importance of the different cellular compartments (endosomes, caveolae) and their essential role in determinating the activity of signaling pathways, also in the case of TGFβ signaling. Endosomal compartments are now generally accepted signaling centers that organize the downstream events and help in the sorting of signal molecules (recycling, ubiquitination, degradation). Emerging new data suggest that early endosomes play important role to define the activity of TGF-β pathways - both (canonical) Smad-dependent and (non-canonical) Smad-independent pathways - as they not only promote the signaling but are important in the termination of the events and can direct the cargos towards degradative pathways. Late endosomal membranes form also a physical surface that can bind the elements of non canonical TGF-β pathways through the lipid compound of caveolae reflecting that both lipid and non-lipid internalization routes are essential for the effective signaling. Endosomal compartments, thus provide a physical platform for the cross-talk of different signaling pathways by binding the singaling molecules that might be the explanation for also the heterogen and complex effects of TGF-β.
To define the exact role of different cytoplasmic compartments with complex integrity of the regulation of canonical and non-canonical pathways might bring us closer to understand better the cellular processes and mechanism of TGF-β induced epithelial-mesenchymal transition.
References
Massagué J (1998) TGF-β signal transduction. Annu Rev Biochem 67:753–91
Shook D, Keller R (2003) Mechanisms, mechanics and function of epithelial-mesenchymal transitions in early development. Mech Dev 120:1351–83
Strutz F, Zeisberg M, Ziyadeh FN, Yang CQ, Kalluri R, Müller GA, Neilson EG (2002) Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int 61:1714–28
Kalluri R, Weinberg RA (2009) The basics of epithelial-mesenchymal transition. J Clin Invest 119:1420–28
Wrana JL, Attisano L, Wieser R, Ventura F, Massagué J (1994) Mechanism of activation of TGF-β receptor. Nature 370:341–47
Shi Y, Massagué J (2003) Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113:685–700
Moustakas A, Souchelnytskyi S, Heldin CH (2001) Smad regulation in TGF-β signal transduction. J Cell Sci 11:4359–69
Zhang Y, Chang C, Gehling JD, Hemmati-Brivanlou A, Derynck R (2001) Regulation of Smad degradation and activity by Smurf2, an E3 ubiquitin ligase. Proc Natl Acad Sci U S A 98:974–79
Tsukazaki T, Chiang TA, Davison AF, Attisano L, Wrana L (1998) SARA, a FYVE domain protein that recruits Smad2 to the TGF-β receptor. Cell 95:779–91
Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana L (2003) Distinct endocytic pathways regulate TGF-β receptor signalling and turnover. Nat Cell Biol 5:410–21
Chen YG (2009) Endocytic regulation of TGF-β signaling. Cell Res 19:58–70
Chen YG, Wang Z, Ma Y, Zhang L, Lu Z (2007) Endofin, a FYVE domain protein, interacts with Smad4 and facilitates transforming growth factor-beta signaling. J Biol Chem 282:9688–96
Gillooly DJ, Simonsen A, Stenmark H (2001) Cellular functions of phosphatidylinositol 3-phosphate and FYVE domain proteins. Biochem J 355:249–58
Itoh F, Divecha N, Brocks L, Brocks L, Oomen L, Janssen H, Calafar J, Itoh S, Dijke PP (2002) The FYVE domain in Smad anchor for receptor activation (SARA) is sufficient for localization of SARA in early endosomes and regulates TGF-beta/Smad signalling. Genes Cells 7:321–31
Le Roy C, Wrana L (2005) Clathrin- and non-clathrin- mediated endocytic regulation of cell signalling. Nat Rev Cell Biol 6:112–26
Bonifacino JS, Lippincott-Schwartz J (2003) Coat proteins: shaping membrane transport. Nat Rev Mol Cell Biol 4:409–14
Bonifacino JS, Traub LM (2003) Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem 72:395–447
Hayes S, Chawla A, Corvera S (2002) TGFβ receptor internalization into EEA1-enriched early endosomes: role in signaling to Smad2. J Cell Biol 158:1239–49
Penheiter SG, Mitchell H, Garamszegi N, Edens M, Dore JJJ, Leof EB (2002) Internalization-dependent and –independent requirements for trasfotming growth factor beta receptor signaling via the Smad pathway. Mol Cell Biol 22:4750–59
Xu L, Chen YG, Massagué J (2000) The nuclear import function of Smad2 is masked by SARA and unmasked by TGFb-dependent phosphorylation. Nat Cell Biol 2:559–62
Heldin CH, Moustakas A (2012) Role of Smads in TGFβ signaling. Cell Tissue Res 347:21–36
Kurisaki A, Kose S, Yoneda Y, Heldin CH, Moustakas A (2001) Transforming growth factor-beta induces nuclear import of Smad3 in an importin-beta1 and Ran-dependent manner. Mol Biol Cell 12:1079–91
Xiao Z, Latek R, Lodish HF (2003) An extended bipartite nuclear localization signal in Smad4 is required for its nuclear import and transcriptional activity. Oncogene 22:1057–69
Mitchell H, Choudhury A, Pagano RE, Leof EB (2004) Ligand-dependent and –independent transforming growth factor-beta receptor recycling regulated by clathrin-mediated endocytosis and Rab11. Mol Biol Cell 15:4166–78
Lisanti MP, Scherer P, Tang ZL, Sargiacomo M (1994) Caveolae, caveolin and caveolin-rich membrane domains: a signalling hypothesis. Trends Cell Biol 4:231–35
Couet J, Li S, Okamoto T, Scherer P, Lisanti MP (1997) Molecular and cellular biology of caveolae: paradoxes and plasticities. Trends Cardiovasc Med 7:103–10
Pelkmans L, Kartenbeck J, Helenius A (2001) Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular transport pathway to the ER. Nat Cell Biol 3:475–83
Nicols BJ (2002) A distinct class of endosome mediates clathrin- independent endocytosis to the Golgi complex. Nat Cell Biol 4:374–78
Parton RG, Simons K (2007) The multiple faces of caveolae. Nat Rev Mol Cell Biol 8:185–94
Kiss AL, Botos E (2009) Endocytosis via caveolae: alternative pathway with distinct cellular compartments to avoid lysosomal degradation? J Cell Mol Med 13:1228–37
Hayer A, Stoaber M, Ritz D, Engel S, Meyer HH, Helenius A (2010) Caveolin-1 is ubiquitinated and targeted to intraluminal vesicles in endolysosomes for degradation. J Cell Biol 191:615–29
Nakao A, Afrakhte M, Moren A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH, PtP D (1997) Identification of Smad7, a TGF-beta inducible antagonist of TGF-beta signalling. Nature 389:631–35
Hayashi H, Abdollah S, Qui Y, Cai J, Xu JJ, Grinnell BW, Richardson MA, Topper JN, Gimbrone MA Jr, Wrana JL, Falb D (1997) The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell 89:1165–73
Zhang S, Fei T, Zhang L, Zhang R, Chen F, Ning Y, Han Y, Feng XH, Meng A, Chen YG (2007) Smad7 antagonizes transforming growth factor beta signaling in the nucleus by interfering with functional Smad-DNA complex formation. Mol Cell Biol 27:4488–99
Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, Wrana JL (2000) Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF-beta receptor for degradation. Mol Cell 6:1365–75
Ebisawa T, Fukuchi M, Murakami G, Chiba T, Tanaka K, Imamura T, Miyazono K (2001) Smurf1 interacts with transforming growth factor-beta type-I receptor through Smad7 and induces receptor degradation. J Biol Chem 276:12477–80
Itoh S, Landström M, Hermansson A, Itoh F, Heldin CH, Heldin NE, PtP D (1998) Transforming growth factor beta1 induces nuclear export of inhibitory Smad7. J Biol Chem 273:29195–201
Kowanetz M, Lönn P, Vanlandewijck M, Kowanetz K, Heldin CH, Moustakas A (2008) TGFbeta induces SIK to negatively regulate type I receptor kinase signaling. J Cell Biol 182:655–62
Bizet AA, Liu K, Tran-Khanh N, Saksena A, Vorstenbosch J, Finnson KW, Buschmann MD, Philip A (2011) The TFG-β co-receptor, CD109, promotes internalization and degradation of TGF-β receptors. Biochim Biophys Acta 1813:742–53
Mukhopadhyay D, Riezman H (2007) Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 315:201–205
Ciechanover A (2005) Proteolysis: from the lysosome to ubiquitin and the proteasome. Mol Cel Biol 6:79–86
Felder S, Miller K, Moshren G, Ullrich A, Schlessinger J, Hopkins CR (1990) Kinase activity controls the sorting of the epidermal growth factor receptor within the multivesicular body. Cell 61:623–34
Gruenberg J, Maxfield F (1995) Membrane transport in the endocytic pathway. Curr Op Cell Biol 7:552–63
von Gersdoff G, Susztak K, Rezvani F, Bitzer M, Liang D, Böttinger EP (2000) Smad3 and Smad4 mediate transcriptional activation of the human Smad7 promoter by transforming growth factor beta. J Biol Chem 275:11320–26
Katzmann DJ, Babst M, Emr DS (2001) Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell 106:145–55
Soond SM, Chantry A (2011) How ubiqitination regulates the TGF-β signalling pathway: new insights and new players. Bioessays 33:749–58
Ito I, Hanyu A, Wayama M, Goto N, Katsuno Y, Kawasaki S, Nakajima Y, Kajiro M, Komatsu Y, Fujimura A, Hirota R, Murayama A, Kimura K, Imamura T, Yanagisawa J (2010) Estrogen inhibits transforming growth factor β signaling by promoting Smad2/3 degradation. J Biol Chem 285:14747–55
Derynck R, Zhang YE (2003) Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 424:577–584
Moustakas A, Heldin CH (2005) Non-Smad TGF-β signals. J Cell Sci 118:3573–3584
Chuderland D, Seger R (2005) Protein-protein interactions in the regulation of the extracellular signal-regulated kinase. Mol Biothechnol 29:57–74
Adachi M, Fukuda M, Nishida E (2000) Nuclear export of MAP kinase (Erk) involves a MAP kinase kinase (MEK)-dependent active transport mechanism. J Cell Biol 148:849–856
Chen RH, Sarnecki C, Blenis J (1992) Nuclear localization and regulation of erk- and rsk-encoded protein kinase. Mol Cell Biol 12:915–927
Kamaraju AK, Roberts AB (2005) Role of Rho/ROCK and p38 MAP kinase pathways in transforming growth factor-β-mediated Smad-dependent growth inhibition of human breast carcinom cells in vivo. J Biol Chem 280:1024–1036
Mori S, Matsuzaki K, Yoshida K, Furukawa F, Tahashi Y, Yamagata H, Sekimoto G, Seki T, Matsui H, Nishizawa M, Fujisawa J, Okazaki K (2004) TGF-β and HGF transmit the signals through JNK-dependent Smad2/3 phosphorylation at the linker region. Oncogene 23:7416–7429
Kretzschmar M, Doody J, Timokhina I, Massagué J (1999) A mechanism of repression of TGF-β/Smad signaling by oncogenic Ras. Genes Dev 13:804–816
Engel M, McDonnell MA, Law BK, Moses HL (1999) Interdependent Smad and JNK signaling in trasforming growth factor-β-mediated transcription. J Biol Chem 274:37413–37420
Kang JS, Liu C, Derynck R (2009) New regulatory mechanisms of TGF-β receptor function. Trends Cell Biol 19:385–394
Galliher AJ, Schiemann WP (2007) Src phosphorylates Tyr284 in TGF-β type II receptor and regulates TGF-β stimulation of p38 MAPK during brest cancer cell proliferation and invasion. Cancer Res 67:3752–3758
Taub N, Teis D, Ebner HL, Hess MW, Huber LA (2007) Late endosomal traffic of epidermal growth factor receptor ensures spatial and temporal fidelity of mitogen-activated protein kinase signaling. Mol Biol Cell 18:4698–4710
Zehorai E, Yao Z, Plotnikov A, Seger R (2010) The subcellular localization of MEK and ERK-A novel nuclear translocation signal (NTS) paves a way to nucleus. Mol Cell End 314:213–220
Zuo W, Chen YG (2009) Specific activation of mitogen-activated protein kinase by transforming growth factor-β receptors in lipid rafts is required for epithelial cell plasticity. Mol Biol Cell 20:1020–1029
Galmiche A, Fueller J, Santel A, Krohne G, Witting I, Doye A, Rolando M, Flatau G, Lemichez E, Rapp UR (2008) Isoform-specific interaction of C_RAF with mitochondria. J Biol Chem 283:14857–14866
Adachi M, Fukuda M, Nishida E (1999) Two co-existing mechanisms for nuclear import of MAP kinase: passive diffusion of a monomer and active transport of a dimer. EMBO J 18:5347–5358
Fukuda M, Gotoh I, Gotoh Y, Nishida E (1996) Cytoplasmic localization of MAP kinase kinase directed by its N-terminal, leucine-rich short amino acid sequence, which acts as a nuclear export signal. J Biol Chem 271:20024–20028
Fukuda M, Gotoh Y, Nishida E (1997) Interaction of MAP kinase with MAP kinase kinase: its possible role in the control of nucleocytoplasmic transport of MAP kinase. EMBO J 16:1901–1908
Acknowledgments
The quality of the manuscript was greatly enhanced by the gracious help of Professor Pál Röhlich. The authors are grateful for his creative and useful scientific comments and the language correction. We are also very grateful for the useful comments and suggestions of the anonymus reviewer(s).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Balogh, P., Katz, S. & Kiss, A.L. The Role of Endocytic Pathways in TGF-β Signaling. Pathol. Oncol. Res. 19, 141–148 (2013). https://doi.org/10.1007/s12253-012-9595-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12253-012-9595-8