
Abstract
The factors involved in the pathogenesis of Crohn’s disease and ulcerative colitis, the two major types of inflammatory bowel disease (IBD) are summarized. Intestinal antigens composed of bacterial flora along with antigen presentation and impaired mucosal barrier have an important role in the initiation of IBD. The bacterial community may be modified by the use of antibiotics and probiotics. The dentritic cells recognize the antigens by cell surface Toll like receptor and the cytoplasmic CARD/NOD system. The balance between Th1/Th2/Th17 cell populations being the source of a variety of cytokines regulates the inflammatory mechanisms and the clearance of microbes. The intracellular killing and digestion, including autophagy, are important in the protection against microbes and their toxins. The homing process determines the location and distribution of the immune cells along the gut. All these players are potential targets of pharmacological manipulation of disease status.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Crohn’s disease (CD) and ulcerative colitis (UC) are the two primary forms of inflammatory bowel disease (IBD). These diseases share many symptoms but in CD, the location of the inflammation may occur anywhere along the digestive tract from the mouth to the anus. In UC, the large intestine (colon) is typically affected; however in some patients the small intestinal ileum also shows the sign of inflammation [1].
Some detail of the relationship between the three items is well known, however, the exact role of the various factors in Crohn’s disease and ulcerative colitis is not well elucidated. In many cases, the two diseases are well differentiated clinically and histologically, but in about 10% of the cases, the transition of the two entities, considered by many as a reasonably separate sub-group, manifests as undetermined colitis.
Immune System of the Intestinal Tract
The immune system of the intestinal wall (gut associated lymphoid tissue: GALT) contains a substantial amount of immune cells responsible for the maintenance of the homeostasis. Due to its villous structure, the gut has a large surface area, the size of a tennis court (260–300 m2), all in contact with the outer environment. Intestinal epithelial cells (IECs) and M-cells lining the gut, therefore have a large area of direct contact with the lumen. Under physiological conditions, the normal intestinal flora of the gut comprises a large number of bacteria, more than the number of somatic cells. There are about 500 species; aerobes and anaerobes live in symbiosis with the host organism. The normal flora protectively impacts the mucosal immune response. It helps protect against pathogens, participates in the metabolism of metabolically active short chain fatty acids, carbohydrates, bile acids and vitamins, and it has an important role in maintaining a balance of the immune system by contributing to the development of a tolerant immune response [2]. The balance of protective and aggressive bacteria seems to be analogous with pro- and antinflammatory cytokine patterns and T-cell sub-populations. The change in the composition of normal flora adversely affects the integrity of the epithelium and the immune protection of mucosa even without the presence of pathogens. Meanwhile, the consequential change in the cytokine composition further impairs the balance. Today, intestinal flora can be investigated or even the precise composition can be analyzed by molecular biological methods; however due to its expensive nature it is not available in routine clinical practice. But it is known that 99% of the bacterial mass comprises only 30–40 species [3].
Ulcerative colitis occurs in sections of the colon where there are the largest numbers of intestinal bacteria. The distal ileum contains 108–1010 bacteria/gram while the colon contains 1011–1012 bacteria/gram, predominantly consisting of aerobes: Bacteroides, Clostridium, Bifidobacteria [4]. Clinical experience, including antibiotic and probiotic therapies, has shown that in animals grown in a germ-free environment no inflammation similar to human IBD occurred suggesting that normal commensal flora has a vital role in bowel inflammation [5].
If IL-10 secreting CD4+ cells together with Th1 helper cells that can cause colitis alone are experimentally transferred into immunodeficient animals, no bowel inflammation occurs due to the antinflammatory effects of IL-10 [6]. It is proposed that in IBD the balance between IL-10 and other cytokines is upset, like other proinflammatory conditions. This can explain the positive effects of antibiotics or probiotics in cases of relapse, which alone is not sufficient and rather “transitory” since the genetic background of the individual does not change and this influences the immune processes as well.
The deficient interaction between bacterial flora and mucosa may lead to an abnormal immune response, inflammation of the intestines, UC and CD. Previous data demonstrates that intestinal flora in IBD is different from a “normal” one [7], but it should be noted that our knowledge of what normal flora is needs to be improved in order to elucidate this issue.
Significance of Immune Tolerance in IBD
The body does not respond with systemic reactions against harmless bacteria and nutritional antigens. The expansion of so-called controlled inflammation is inhibited by CD4+ T-cells [8]. Processes include T-cell anergy and apoptosis as well as central clone deletion. Additional suppressor mechanisms include the release of T-cell induced inhibitory cytokines: IL-10, TGF-β, IL-4 [9].
T-cell apoptosis has a critical role in immune homeostasis. In normal mucosa there is a high rate of Fas-mediated apoptosis in T-cells of the lamina propria. However, in both CD and UC the level of apoptosis is found to be significantly decreased [10]. In patients with CD the level of Bcl-2 and Bcl-xl with anti-apoptotic activity is increased in T-cells, while Bax protein stimulating apoptosis is decreased resulting in slower apoptosis [11].
Many experimental and human data demonstrate that inflammatory bowel disease is the consequence of the lack of normal mucosal immune tolerance. In IBD the ratio of antibodies produced against nutritional antigens is elevated. In the absence of appropriate tolerance, the usual luminal flora induces active, non-regulated immune response. The immune cells of the intestinal wall loose the capability to differentiate between potential pathogens and non-pathogenic bacterial environment. Oral tolerance can be stimulated by food intake similarly to that occurring in active immunization as in case of Sabin drops [12]. This method was shown to be useful both in the prevention and therapy of IBD [13, 14]. By local application of trinitrobenzoic acid (TNBA) Crohn-like, IL-12 mediated Th1 type colitis can be provoked in mice. In this case, the preventive effect of antigen feeding could be demonstrated [15]. This phenomenon could not be similarly reproduced in humans, indicating that regarding oral tolerance there is a fundamental difference between animal and human colitis.
With this in mind several attempts have partially succeeded in demonstrating the differences of oral tolerance among patients with inflammatory bowel disease, their relatives and the normal population [16]. Although various theories exist regarding the pathomechanism of IBD, based on animal models, researchers agree that the intake of the appropriate antigen could attenuate the inflammatory process. The best findings have been reported by Ilian at al. [17]. Colon biopsy samples were taken from ten patients with CD, prepared and administered as food. After sixteen weeks, significant decrease in CDAI was observed, furthermore the peripheral NKT cell ratio increased, serum IL-4 and IL-10 levels were elevated demonstrating activation of the antinflammatory system.
A similar mechanism can be proposed for how probiotics and prebiotics work in routine practice; positive results are achieved by influencing oral tolerance. Beneficial therapeutic effects described in animal and human pouchitis look to be promising [18]. Nevertheless, results are contradictory and there is reasonable doubt. One explanation could be the lack of appropriate antigen, dose and the duration of treatment.
Antigen Presentation at the Mucosal Barrier
Dendritic cells, significant elements of antigen presentation, reside in the luminal surface of mucosa and perceive antigens in the intestinal content via their dendrites. In IBD, their number is increased compared to normal and expression of MHCII and T-cell co-stimulation molecules on their surface is enhanced. An increase in interferon-γ, IL-12, IL-18 release from dendritic cells (IL-4, IL-13 remain unchanged) can be observed in a mice model compared to healthy controls. There are also larger numbers of dendritic type cells in the mucosa associated lymphoid tissue of patients with CD [19].
The functional condition of the predominantly myeloid origin antigen presenting cells (monocytes and macrophages) in lamina propria depends on their location, maturity and the degree of inflammation [20]. In the murine Peyer’s patch, two different populations of dendritic cells (DC) are localized: in the subepithelial dome myeloid and in the interfollicular regions lymphoid ones [21]. If CD11+ dendritic cells are isolated from normal human colon previously treated with a prebiotic agent (VSL#3), increased IL-10 and decreased IL-12p40 production (i.e. the antinflammatory processes) will dominate. At the same time, lipopolysaccharide stimulated IL-12p40 production can be stopped by adding VSL#3 [22]. It is important that the commensal flora continues to live in the dendritic cells of Peyer’s patches and mucosal lymphoid foci, but from here it does not penetrate further and cannot be obtained from the spleen. Further penetration is inhibited by an IgA response independent from T-cell line [23].
Dendritic cells present intestinal antigens to naïve T-cells of the secondary lymphoid system (Peyer’s patches and mesenteric lymph nodes). If dendritic cells obtained from peripheral lymph node, spleen and Peyer’s patches are co-incubated with CD8+ T-cells, the markers indicating activation will increase irrespective of the site of origin. However, only dendritic cells from Peyer’s patches are able to increase the expression of adhesion factors (integrin α4β7) and enhance “homing” of T-cells (i.e. increasing the probability of encountering with the antigen) [24]. These observations demonstrate that dendritic cells have a significant role not only in recognition of the antigen but also in the intestinal migration of T-cells. Based on the above, it is not surprising that dendritic cells have a key role in oral tolerance. Experimental observations dictate that provoking expansion of the dendritic cells results in increase of tolerance.
In addition to the theoretical approach, observations also confirm the role of these cells in IBD. Experimental models demonstrate changing of dendritic cells in inflammatory conditions. For example in the CD45RBhi colitis model an increase of CD134L+ dendritic cells was found in the mucosal lymphoid tissue following pathogenic T-cell transmission in RAG1−/− mouse [25]. There is much less data available regarding human IBD. Bell et al. have found CD40, CD80, CD83 and CD86 co-stimulator molecule expression in CD11+ dendritic cells isolated from rectum and colon histological samples from patients with CD. These results correlate with previous immunohistological observations [26].
Others described in CD an altered behavior of dendritic cells and the lack of thymic stromal lymphopoietin production stimulating their maturity [27]. The importance of this topic is emphasized by the fact that dendritic cells have a significant role in how the innate and adaptive immune cells react with the external environment, the maintenance of homeostasis, and the development of a tolerant or eliminating immune response. Does the dendritic cell, having several subtypes, function well in IBD or does the erroneous function have more of a role in the development of the disease? Answers to these questions may open the way to new therapeutic alternatives.
The Role of Innate Immune System in Perception of Antigen: NOD and Toll-like Receptors
Discovery of the antigen recognizing structures of the innate immune system (PRR—pattern recognition receptor) are significant advances of the last decade. These receptors are expressed on various cell types and the elements of this system represent the first line in the microbial-mucosal interaction and this process fundamentally affects further events [28].
Toll like receptors (TLRs) are sensors on the cellular surface, following contact with antigens they initiate proinflammatory cytokine expression through the NF-κB system that is the link between innate and adaptive immunity. Deficiency or malfunctioning of these sensors may lead to loss of adequate control of inflammatory processes leading to the development of IBD.
The 14 (human and mouse) TLRs are categorized into 5 main groups, but their number is expected to increase in the future. Many studies have demonstrated increased TLR expression in IBD mucosa compared with healthy controls [29]. There is the most data regarding the first described TLR4 expressed typically on the apical surface of the cells. Nearly 50% of the intestinal flora comprises Gram-negative bacteria. The main antigen of gram negative bacteria is a lipopolysaccharide (LPS) that serves as a substrate for the TLR4 receptor. Another molecule called MD-2 participates in recognition of the antigen; the expression of this can hardly be measured in the normal colon mucosa, while in IBD it is significantly elevated. The increased expression of TLR4, MD-2, TNF-α, interferon-γ observed in the inflamed intestine is the result of over-response to the bacterial lipopolysaccharide [30]. TLR4 expression is not uniform in the small and large intestines, possibly because of the differing bacterial flora [31].
The possible significance of TLR4 in IBD is explained by its specific behavior regarding the bacterial flora. It is the most sensitive receptor to LPS to an extent that this feature if it can be experimentally transmitted into animals and the decreased LPS signal can be restored [32]. Activation of TLR4 results in expression of NF-κB, myd88 protein, MAP kinases, and nuclear receptor peroxisome proliferator activated receptor gamma (PPAR-γ) [33]. TLR2 is typically expressed in the proximal colon and under experimental conditions chemically induced colitis models. Immunohistochemical analysis of crysections showed a significant increase in TLR2 expression in the terminal ileum of patients with inactive and active UC against controls. In the same study significant upregulation of TLR4 expression was found in the terminal ileum and rectum of UC patients in remission and in the terminal ileum of CD patients with active disease. CD14 expression was up-regulated in the terminal ileum of CD patients in remission and with active disease, in the cecum of UC patients in remission and with active disease, and in rectum of UC patients with active disease. Hence, dysregulation of TLR2, TLR4, and CD14 expression in different parts of the intestinal mucosa may be crucial in IBD pathogenesis [34]. The most recent findings emphasized the role of lipopolysaccharide-binding protein and soluble CD14 in CD [35]. Epidemiological data have indicated that occurrence of childhood infections is in inversely proportional to the risk of developing CD [36]. In vitro and in vivo studies can confirm that bacterial LPS and CpG DNA enhances Th1 response via IL-12 produced by dendritic cells [37]. Expression of TLR3 was found to be lower in patients with CD when compared with those with UC and healthy controls [38]. TLRs are not only expressed in the intestinal epithelium but also in macrophages of the lamina propria, the intestinal myofibroblasts, and the endothelial cells of capillaries indicating their key role in innate immunity. It is likely that genetic polymorphisms of the TLR receptor affect the sensitivity of response to the pathogenic flora.
The protein family of NOD system located in the cytosol is the receptor of innate immunity similar to TLR, and participates in the reaction between host and pathogens. It is beyond doubt that the most exciting discovery of the recent period has been identifying the role of NOD2/CARD15 gene in IBD. The NOD2 protein has a significant part in the development of the inflammatory response against bacterial proteins (LPS and heat shock proteins) and its genotype is associated with seroreactivity to microbial components in CD [39]. The bacterial antigen attached to the Toll-like receptor on the surface of monocytes and macrophages and the cytoplasmic NOD2 protein induces the corresponding pro- and antinflammatory cytokines via NF–κB [40]. So it is not surprising, that NF–κB genetic variant is associated with extensive colitis in IBD patient [41]. According to our current knowledge, the role of NOD2/CARD gene is markedly different in CD and UC. In the latter, no significant correlation has been found between gene mutations and developing the diseases, while in CD this is confirmed by many studies [42]. This observation is in accord with previous studies that have shown that a bacterial event (infection) may have an inductive role in the development of CD more so than in ulcerative colitis.
NOD2 mutations occur more often in stenotic Crohn patients [43, 44]. This discovery has brought about a marked advancement in the investigation of the pathomechanism of UC. This is the first indication of the significance of genetics and focused researchers attention on the role of bacterial “crosstalk” in disease etiology. Mutation occurs in the leucine-rich part of the NOD2 molecule. This area is responsible for recognition of a muramyl-dipeptide, the smallest part of the peptidoglycan maintaining its antigenic features [45, 46]. In Crohn colitis, NOD2 expression of epithelial cells is increased compared to normal [47]. Proinflammatory cytokines such as TNF-α and IFN-γ are able to increase NOD2 expression. In vitro studies have demonstrated higher NOD2 expression of intestinal epithelial cells against anti-bacterial pathogens such as Salmonella typhimurium [48].
CARD15/NOD2 mutations are neither necessary nor sufficient for disease occurrence. This observation is in accordance with the retained complex genetic model for CD, where genetic risk factors contribute only to a small proportion of the variance (CARD15/NOD2 mutations are very rare in Asian or African populations). The risk of disease may appear relatively high in the people with mutations on two of the two chromosomes. This dosage effect is consistent with a loss-of-function model, where the molecular defect of the gene lowers its efficiency. This loss-of-function model was confirmed by many data from several groups using in vitro models of transiently transfected cell lines [44].
In contrast, the gain-of-function mutations of CARD15/NOD2 was also proposed based on potentiation of NF-κB activity and IL-1β processing in mouse model. CARD15/NOD2 is able to activate IL1β via the caspase 1 pathway confirms the proinflammatory properties of CARD15/NOD2 [49].
NOD1 is specialized to the peptidoglycan-muramyl tripeptide of Gram-negative bacteria.
Experiments demonstrate that in NOD1-deficient mouse bacteria are not eliminated due to the lack of response against them. NOD1 system has important role in the elimination of enteroinvasive, Gram-negative bacteria, like Shigella flexneri [50].
Intestinal expression of NOD proteins is the manifestation of the reaction against the bacterial environment. When it is deficient, function bowel inflammation may develop, so it is not surprising that NOD2 mutations can be detected in some patients with CD.
NOD2 expression can be observed not only on immune cells but also on Paneth-cells, justifying paying increased attention to this subject [51]. Paneth-cells are located at the base of Lieberkuhn crypts and have a special role in the protection of epithelial surface by producing antimicrobial molecules. This occurs in large numbers mainly in the terminal section of the ileum. In NOD2 mutant mice, Paneth-cells produce significantly less antimicrobial peptides (cryptidins) and the protection against orally administered bacteria is decreased. This corresponds with the observation that bacterial adherence in the terminal ileum of patients with CD, which is associated with the lack of adequate protective apparatus (defensins), is significantly increased compared to the normal population. Antimicrobial activity of defensins and cathelicidins is required for the protection of the integrity of normal mucosa. In the small intestine, α-defensin of the Paneth-cell is able to regulate microorganisms, while β-defensin and cathelicidin may induce an inflammatory reaction [52] (Figs. 1 and 2).

The factors of antigen presentation leading to inflammatory bowel disease syndrome. Th: T helper cell, IgA: immunoglobulin A, PG: peptidoglycans, LPS: lipopolysaccharides, CpG: CpG dinucleotides, TLR: Toll like receptors, APC: antigen presenting cell, NOD: nucleotide binding domain

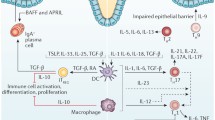
The major regulatory factors in the pathogenesis of inflammatory bowel disease (IBD). The arrows show the enhancement and the blunt end symbols represent the inhibition. IL: interleukin, TGF: transforming growth factor, MMPs: matrix metalloproteinases
Any defects or damage to the barrier allow intrusion of bacterial antigens leading to chronic inflammation. Precise understanding of the role of defensins may provide the key to a new therapeutic method, which could be tested through administration of probiotics.
Polarization of the Adaptive Immune Response: Th1 / Th2
CD is characterized by Th1 dominance, while in UC is the less typical excess of Th2 dominates [53].
Altered composition of T-cells present in the lamina propria can be typical for CD, and UC versus healthy controls. In CD, CD4+ CD45R0+ T-cells and CD68 positive macrophages dominate. CD4+ T-cells are predominantly of the Th1 phenotype and produce TNF-α, IFN-γ and IL-6 [54]. In UC, IL-5 dominates in the cytokine profile and to a lesser extent IFN-γ, which is characteristic of the Th2 phenotype [55]. T-cells (CD4+ or CD25+) producing regulating antinflammatory cytokines against T-cell production of Th1 and Th2 type proinflammatory cytokines, and Th1 producing IL-10 and Th3 producing TGF-β are found in the intestinal wall [56]. In CD T-cell activation may be a result of IL-12, IL-23 and IL-18 cytokines released from dendritic cells and macrophages by encountering luminal antigens [57]. Interferon-γ produced subsequent to initiation of the process makes it self-maintaining through intracellular STAT1-4 and T-bet proteins [58, 59]. In UC, one of the key inductive cytokines may be IL-13. [60]. Expression of other cytokines and proteins have been described, among them perhaps IL-27, Ebi-3, p28 subunit should be mentioned; however the exploration of novel signal pathways may also be predicted.
In addition to other mechanisms, the cytokine milieu is also characterized by the excessive presence of anti-apoptotic ones: IL-2, IL-6, IL-15, IL-17, and IL-18. During anti-TNF-α treatment, it was observed that while infliximab with strong apoptotic potential worked well, while etanercept without such an effect failed. Apoptotic activity is also important in other treatments used for IBD, such as the immunosuppressive drugs azathioprine and sulfasalazine.
The success of anti-TNF-α treatment in UC may also be due to the apoptotic activity or the modulation of IL-33/ST2 system which plays an important role in IBD and experimental colitis, and may represent a specific biomarker for active UC [61]. Upon introduction of infliximab, UC was considered a contra-indication because the main effect of the active substance was considered to block Th1 released TNF-α. Experience with other active substances (etanercept) and the success of Phase 2 studies in UC focused researchers’ attention to a common dominant factor in maintenance of chronic inflammation; the lack of apoptosis, characteristic to both types of IBD.
The effects of intercellular cytokines develop through intracellular messengers targeting transcription factors. One of the intracellular signal transducers is the STAT system, which plays role in the post receptor signaling of different antinflammatory cytokines, activated by the IL-6 cytokine family, IL-10, G-CFS, and hepatocyte growth factor [62]. This is significant because in IBD the endogenous inhibitor (SOCS3) level is increased, reducing the efficiency of antinflammatory cytokines and enhancing apoptosis resistance [63]. Experimental data have shown that STAT3 deficiency leads to IL-10 functional disorder and severe gastrointestinal inflammation.
TGF-β also has antinflammatory potential. Under experimental conditions, its deficiency causes severe bowel inflammation or death [64]. Signal transduction of TGF-β is mediated by Smad 2 protein, and the level of its natural inhibitor, Smad7, is increased in IBD which also has a proinflammatory effect. This is the explanation for the increased TGF-β levels in IBD but without antinflammatory effects due to the lack of an adequate signal transducer [65].
Deficient innate immunity results in faulty T-cell response; and in such conditions (chronic granulomatosis, glycogen storage disorders, cyclic neutropenia and leukocyte adhesion syndrome) “crohn”-like intestinal alterations may be observed [66]. Therefore it is postulated that stimulation of innate immunity may have therapeutic effect in patients with CD.
Role of TH17 Cells
TH17 (CD4+) T-cell lineage has been is characterized by the production of the cytokine IL-17 and the development of which is promoted by IL-23 and suppressed by transcription factors required for TH1 and TH2 cells [67, 68]. It now appears that IL-23 is unnecessary for the initiation of Th17 differentiation but is required at a crucial control point in the Th17 response. STAT3, which is activated by IL-23, was shown to be essential in expansion and/or maintenance of the Th17 response. [69]. In human T-cells, IL-23, together with TGF-β, IL-1β and IL-6 were shown to induce Th17 differentiation and expression of IL-17A, IL-17F, the IL-23R and the transcription factor RORγt which is required for Th17 differentiation [70]. IL-23 was shown to suppress expression of Foxp3 which induces regulatory T-cell development IL-23 was shown to suppress expression of Foxp3 which induces regulatory T-cell development [71]. If Foxp3 is present in T-cells, it can divert differentiation away from Th17 because it binds RORγt and blocks its action. Since Foxp3+RORγt+ cells are found in the gut associated lymphoid tissue, it has been suggested that IL-23 acts on this intermediate cell, suppressing Treg development and thereby promoting Th17 development [72].
So the TH17 cell lineage commitment is driven by TGF-β in the presence of proinflammatory cytokines, whereas IL-23 seems to maintain TH17 cell populations [73]. In addition to its ability to support the differentiation of TH17 cells, IL-23 induces the secretion of IL-17 by non-T-cells in an inflammatory environment, and both T-cells and monocytes serve as sources of increased expression in the mucosa of IBD patients. IL-17 may induce antimicrobial peptides and may regulate tight junction barrier formation [74].
The TH17 cells produce IL-17A, IL-17F and IL-22 besides other cytokines. Th17 cells are clearly capable of inducing intestinal inflammation, it remains to be determined which Th17 cell effector cytokines actually mediate the inflammatory response. One candidate for pathogenesis by Th17 cells is IL-17F. Although IL-17A and IL-17F have overlapping activities in some systems, there is evidence for differential activities in the gut. [70]
Intracellular Digestion and Autophagy
The impared phagosomal functions may reduce the pathogen elimination and handling of commensal flora. An SNP within the first intron of neutrophil cytosol factor 4 (NCF4/p40Phox) is associated with enhanced susceptibility to CD. The protein product of this gene is a cytosolic regulatory component of the superoxide-producing phagocyte NADPH-oxidase [75]. Functionally impaired phagosomes are less effectively in the killing resulting prolonged microbe elimination and immune activation [76].
The autophagy pathway plays role in protecting mammalian cells against various the cytotoxic effect of bacterial toxins [77]. Following pathogen infection, autophagy could represent a primary attempt to re-establish homeostasis together with apoptosis. The relevance to IBD is highlighted by the recent discovery that an SNP in the auto-phagocytic gene ATG16L1 is associated with increased risk for Crohn’s disease [78, 79]. ATG16L1 is broadly expressed in the intestinal epithelium, APCs, T- and B-cells and may be involved in the host response to intracellular bacteria [75]. Another autophagy gene IRGM, required for mycobacterial elimination, is also associated with Crohn’s disease risk [80], and may have an analogous role in the granulomatous response observed in Crohn’s disease [81, 82].
Not only intracellular digestion, but extracellular one by matrix metalloproteases is also suggested factor and potential differential diagnostic tools for IBDs [83].
Lymphocyte “Homing”
“Lymphocyte homing” phenomenon is worthy to note for pathophysiological and therapeutic reasons. During an immune reaction due to antigen stimulus, the sensitized T-cell should move from the central immune organ to the periphery, into the target organ. This is usually the gut, but in case of extra-intestinal manifestations, alteration of homing in the endothelium will determine the target organ. After encounter with the antigen, (priming) return of effector cells into the lamina propria (homing) depends on the expression of adhesion molecules. In IBD, the expression pattern of adhesion molecules changes. On the surface of CD4+ T-cells β2 integrin LFA1, α4β1 and α4β7 integrins are expressed, while on the surface of the endothelial cell ICAM-1, VCAM-1 and MadCAM-1 appear [84]. Both LFA1 and ICAM-1 expression is increased in IBD compared to healthy control. Thus, modulation of expression of adhesion molecules may inhibit the elements maintaining the inflammation from reaching the lamina propria that may have beneficial therapeutic effect [85]. The role of Peyer’s patch derived dentritic cells in the regulation of T-cell α4β7 integrin expression and lymphocyte homing was discussed in the section about antigen presentation.
Conclusion
Based on the diverse and in part controversial experimental observations and scientific publications, it is difficult to draw conclusions regarding the pathomechanism of IBD. Nevertheless, the significantly genetically determined innate immunity seems to be of primary importance in IBD, and in particular in Crohn’s disease. Consequently, when there is damage to the communication between the intestinal flora and the immune system locally uncontrolled inflammation is likely. Additionally, genetic background determines the uncontrolled immune reactions that function abnormally and thus are unable to correct the deficiency. It is associated with the damage of mucosal barrier, continuous, unlimited intrusion of invasive strains into the deeper structures that maintain chronic inflammation due to the immune response; at this stage considered normal. It is hard to determine if immunological imbalance is the cause or the outcome, but knowing the “double-face” of the system it is likely that both factors are present at the same time. The condition that is well known is caused by the hyperactivity of the sensitized system due to genetic predisposition. The degree of self-correction is likely to be genetically regulated as well. An example of how this is likely, is the efficiency of probiotics in pouchitis where administration of antigen/probiotics alone has therapeutic effects. This observation calls attention to another essential issue: is the alteration of the bacterial environment in IBD a result of deficient function of the immune system or does a change in the bacterial flora (for unknown reasons) cause chronic, uncontrolled inflammation in genetically sensitized animals. These are unanswered questions, but with the increased rate of research in this area the numbers of such questions will more than likely decrease in future.
Up to now, several factors have been proposed to be involved in the pathogenesis of IBD, including bacterial antigens of the intestinal flora and anomalies of immune regulation, phagocytosis, antigen presentation (Fig. 1) and balance between the different T-cell subpopulations (Fig. 2). The various manipulation of these factors by chemotherapy or probiotics, may lead to the prevention and therapy of this severe pathologic condition in the future.
Abbreviations
- APC:
-
antigen presenting cell
- ATG16L1:
-
autophagy-related protein 16–1
- CARD:
-
caspase recruitment domain-containing protein
- CD:
-
Chron’s disease
- CD:
-
cluster of differentiation
- CDAI:
-
crohn’s disease activity index
- CpG:
-
cytosine—phosphate—guanine
- DC:
-
dendritic cell
- DSS:
-
dextran sulphate sodium
- Ebi:
-
Epstein-Barr virus induced gene
- GALT:
-
gut associated lymphoid tissue
- G-CSF:
-
granulocyte colony stimulating factor
- IBD:
-
inflammatory bowel disease
- ICAM:
-
intercellular adhesion molecule
- IEC:
-
intestinal epithelial cells
- Ig:
-
immunoglobulin
- IL:
-
interleukin
- INF:
-
interferon
- IRGM:
-
immunity-related GTPase family M
- KGF:
-
keratinocyte growth factor
- KSR:
-
Ras-1 kinase suppressor
- LFA:
-
lymphocyte function-associated antigen
- LPS:
-
lipopolysaccharide
- MHC:
-
major histocompatibility complex
- MMP:
-
matrix metalloprotease
- NADPH:
-
nicotinamide adenine dinucleotide phosphate
- NCF4:
-
neutrophil cytosol factor
- NF:
-
nuclear factor
- NKT:
-
natural killer T cell
- NLR:
-
NOD type receptor
- NOD:
-
nucleotide-binding oligomerization domain protein
- PAMP:
-
pathogen associated molecular pattern
- PAR:
-
proteinase activated receptor
- PPAR:
-
peroxisome proliferator activated receptor
- PRR:
-
pattern recognition receptor
- RAG:
-
recombination-activating gene
- Ras:
-
rat sarcoma oncogene
- SCID:
-
severe combined immunodeficiency
- Smad:
-
vertebrate homologue of Mad (Mothers Against Decapentaplegic) protein
- SOCS:
-
suppressor of cytokine signaling
- STAT:
-
signal transducers and activators of transcription
- TGF:
-
transforming growth factor
- Th:
-
T helper
- TLR:
-
toll-like receptor
- TNBA:
-
trinitrobenzoic acid
- TNF:
-
tumor necrosis factor
- TNFR:
-
TNF receptor
- TRC:
-
T cell receptor
- UC:
-
ulcerative colitis
- VCAM:
-
vascular cell adhesion molecule
References
Tresca AJ (2009) Differences Between Ulcerative Colitis and Crohn’s Disease. In: About.com: Health. http://ibdcrohns.about.com/od/ulcerativecolitis/a/diffuccd.htm. Cited 16. Jun 2010
Balfour SR (2001) Induction of mucosal immune responses by bacteria and bacterial components. Curr Opin Gastroenterol 17:555–561
Mai V, Morris JG (2004) Colonic bacterial flora: changing understandings in the molecular age. J Nutr 134:459–464
Wilson KH (2002) Natural biota of the human gastrointestinal tract. In: Blaser MJ, Smith PD, Ravdin JI (eds) gastrointestinal tract. Lippincott Williams & Wilkins, Philadelphia
Danese S, Sans M, Fiocchi C (2004) Inflammatory bowel disease: the role of environmental factors. Autoimmun Rev 3:394–400
Cong Y, Weaver CT, Lazenby A et al (2002) Bacterial-reactive T regulatory cells inhibit pathogenic immune responses to the enteric flora. J Immunol 169:6112–6119
Bai PA, Ouyang Q (2006) Probiotics and inflammatory bowel disease. Postgrad Med J 82:376–382
Groux H, O'Garra A, Bigler M et al (1997) A CD4+ T-cell subset inhibits antigen specific T-cell responses and prevents colitis. Nature 389:737–742
Abbas AK, Murphy KM, Sher A (1996) Functional diversity of helper T lymphocytes. Nature 383:787–793
Boirivant M, Marini M, Di Felice G et al (1999) Lamina propria T-cells in Crohn’s disease and other gastrointestinal inflammation show defective CD2 pathway-induced apoptosis. Gastroenterology 116:557–565
Bu P, Keshavarzian A, Stone DD et al (2001) Apoptosis:one of the mechanisms that maintains unresponsivness of the intestinal mucosal immune system. J Immunol 166:6399–6403
Nagler-Anderson C, Bober LA, Robinson ME et al (1986) Suppression of type II collagen- induced arthritis by intragastric administration of soluble type II. collagen. Proc Natl Acad Sci USA 83:7443–7446
Higgins PJ, Weiner HL (1988) Suppression of experimental encephalomyelitis by oral administration of myelin basic protein and its fragments. J Immunol 140:440–445
Vrabec TR, Gregerson DS, Dua HS et al (1992) Inhibition of experimental autoimmun uveoretinitis by oral administration by oral administration of S-antigen and syntetic peptides. Autoimmunity 12:175–184
Neurath MF, Fuss I, Kelsall BL et al (2004) Experimental granulomatous colitis in mice abrogated by induction of TGF-β mediated oral tolerance. J Exp Med 183:2605–2616
Kraus TA, Toy L, Chan L et al (2004) Failure to induce oral tolerance in Crohn’s disease and ulcerative colitis patients: posible genetic risk. Ann NY Acad Sci 1029:225–238
Ilian Y (2004) Oral immune ragulation toward disease-assiciated antigens: results of phase I clinical trials in Crohn’s disease and chronic hepatitis. Ann NY Acad Sci 1029:286–298
Hart AL, Stagg AJ, Kamm MA (2003) Use of probiotics in the treatment of inflammatory bowel disease. J Clin Gastroenterol 36:111–119
Niess JH (2008) Role of mucosal dendritic cells in inflammatory bowel disease. World J Gastroenterol 14:5138–5148
Coombes JL, Maloy KJ (2007) Control of intestinal homeostasis by regulatory T-cells and dendritic cells. Semin Immunol 19:116–126
Iwasaki A, Kelsall BL (2001) Unique functions of CD11b+, CD8 alpha+, and double-negative Peyer’s patch dendritic cells. J Immunol 166:4884–4890
Hart AL, Lammers K, Brigidi P et al (2004) Modulation of human dendritic cell phenotype and function by probiotic bacteria. Gut 53:1602–1609
Macpherson AJ, Uhr T (2004) Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science 303:1662–1665
Mora JR, Bono MR, Manjunath N et al (2003) Selective imrprinting of gut-homing T cells by Peyer’s patch dendritic cells. Nature 424:88–93
Krajina T, Leithäuser F, Möller P et al (2003) Colonic lamina propria dendritic cells in mice with CD4+ T cell-induced colitis. Eur J Immunol 33:1073–1083
Bell SJ, Rigby R, English N et al (2001) Migration and maturation of human colonic dendritic cells. J Immunol 166:4958–4967
Rimoldi M, Chieppa M, Salucci V et al (2005) Intestinal immun homeostasis is regulated by the crosstalk between epithelial cells and dendritic cells. Nat Immunol 6:507–514
Xavier RJ, Podolsky DK (2007) Unravelling the pathogenesis of inflammatory bowel disease. Nature 448:427–434
Suzuki M, Hisamatsu T, Podolsky DK (2003) Gamma interferon augments the intracellular pathway for lipopolysaccahride (LPS) recognition in human intestinal epithelial cells through coordinated upregulation of LPS uptake and expression of the intracellular Tolle-like receptor 4-MD-2 complex. Infect Immun 71:3503–3511
Ortega-Cava CF, Ishihara S, Rumi MA et al (2003) Strategic compertmentalization of toll-like receptor 4 in the mouse gut. J Immunol 170:3977–3985
Abreu MT, Vora P, Faure E et al (2001) Decreased expression of Toll-like receptor-4 and MD-2 correlates with intestinal epithelial cell protection adainst dysregulated proinflammatory gene expression in response to bacterial lipopolysaccharide. J Immunol 167:1609–1616
Cario E, Podolsky DK (2000) Differencial alteration in intestinal epithelial cell expression of toll like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun 68:7010–7017
Dubuquoy L, Jansson EA, Deeb S et al (2003) Impaired expression of peroxisome proliferator-activated receptor gamma in ulcerative colitis. Gastroenterology 124:1265–1276
Frolova L, Drastich P, Rossmann P et al (2008) Expression of Toll-like receptor 2 (TLR2), TLR4, and CD14 in biopsy samples of patients with inflammatory bowel diseases: upregulated expression of TLR2 in terminal ileum of patients with ulcerative colitis. Histochem Cytochem 56:267–274
Lakatos PL, Kiss LS, Palatka K et al (2011) Serum lipopolysaccharide-binding protein and soluble CD14 are markers of disease activity in patients with Crohn’s disease. Inflamm Bowel Dis 17:767–777
Trinchieri G (2003) Interleukin-12 and regulation of innate resistance and adaptive immunity. Nat Rev Immunol 3:133–146
Mosmann TR, Cherwinski H, Bond MW et al (1986) Two types of murine helper T cell clone. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol 136:2348–2357
Hayashi F, Smith KD, Ozinsky A et al (2001) The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410:1099–1103
Papp M, Altorjay I, Norman GL et al (2007) Seroreactivity to microbial components in Crohn’s disease is associated with ileal involvement, noninflammatory disease behavior and NOD2/CARD15 genotype, but not with risk for surgery in a Hungarian cohort of IBD patients. Inflamm Bowel Dis 13:984–992
Abreu MT, Taylor KD, Lin YC et al (2002) Mutations in NOD2 are associated with fibrostenosing disease in patients with Crohn’s disease. Gastroenterology 123:679–688
Szamosi T, Lakatos PL; Hungarian IBD Study Group et al (2009) The 3′UTR NFKBIA variant is associated with extensive colitis in Hungarian IBD patients. Dig Dis Sci 54:351–359
Hugot JP, Chamaillard M, Zouali H et al (2001) Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411:599–603
Chamaillard M, Philpott D, Girardin SE (2003) Gene enviroment interaction modulated by allelic heterogenety in inflammatory bowel diseases. Proc Natl Acad Sci USA 100:3455–3460
Hugot J-P (2006) CARD15/NOD2 mutations in Crohn’s disease. Ann NY Acad Sci 1072:9–18
Abreu MT, Arnold ET, Thomas LS et al (2002) TLR4 and MD-2 expression is regulated by immune medaited signals in human epithelial cells. J Biol Chem 277:20431–20437
Weigmann B, Nemetz A, Becker C et al (2004) A critical regulatory role of leucin zipper transcription factor c-Maf in Th1-mediated experimental colitis. J Immunol 173:3446–3455
Elson CO, Cong Y, Iqbal N et al (2001) Immuno-bacterial homeostasis in gut: new insight into the old enigma. Semin Immunol 13:187–194
Boirivant M, Fuss IJ, Chu A et al (1998) Oxazolone colitis: a murine model of T helper cell type 2 colitis treatable with antibodies to interleukin 4. J Exp Med 188:1929–1939
Maeda S, Hsu LC, Liu H et al (2005) Nod2 mutation in Crohn’s disease potentiates NF-kappaB activity and IL-1beta processing. Science 307:734–738
Girardin SE, Boneca IG, Carneiro LA et al (2003) Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 300:1584–1587
Lala S, Ogura Y, Osborne C et al (2003) Crohn’s disease and the NOD2 gene: a role for paneth cells. Gastroenterology 125:47–57
Ramasundara M, Leach ST, Lemberg DA, Day AS (2009) Defensins and inflammation: the role of defensins in inflammatory bowel disease. J Gastroenterol Hepatol 24:202–208
Inohara N, Ogura Y, Fontalba A et al (2003) Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem 278:5509–5512
Berrebi D, Maudinas R, Hugot JP et al (2003) Card15 gene overexpression in mononuclear and epithelial cells of the inflammed Crohn’s disease colon. Gut 52:840–846
Hisamatsu T, Suzuki M, Reinecker HC (2003) CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology 24:1001–1009
Brimnes J, Reimann J, Nissen M et al (2001) Enteric bacterial antigens activate CD4(+) T cells from scid mice with inflammatory bowel disease. Eur J Immunol 31:23–31
Peluso I, Pallone F, Monteleone G (2006) Interleukin-12 and Th1 immune response in Crohn’s disease: pathogenetic relevance and therapeutic implication. World J Gastroenterol 12:5606–5610
Neurath MF, Weigmann B, Finotto S et al (2002) The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Cronh’s disease. J Exp Med 195:1129–1143
Afkarian M, Sedy JR, Yang J et al (2002) T-bet is a STAT1-induced regulator of IL-12R expression in naïve CD4+ T cells. Nat Immunol 3:506–508
Heller F, Fuss IJ, Nieuwenhuis EE et al (2002) Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL-13-producing NK-T cells. Immunity 17:629–638
Pastorelli L, Garg RR, Hoang SB et al (2010) Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proc Natl Acad Sci U S 107:8017–8022
Levy DE, Lee CK (2002) What does Stat3 do? J Clin Invest 109:1143–1148
Mudter J, Weigmann B, Bartsch B et al (2005) Activation pattern of signal transducer and activators of transcription (STAT) factors in inflammatory bowel diseases. Am J Gastroenterol 100:64–72
Shull MM, Ormsby I, Kier AB et al (1992) Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature 359:693–699
Babyatsky MW, Rossiter G, Podolsky DK (1996) Expression of transforming growth factors alpha and beta in colonic mucosa in inflammatory bowel disease. Gastroenterology 110:975–984
Yamaguchi T, Ihara K, Matsumoto T et al (2001) Inflammatory bowel disease-like colitis in glycogen storage disease type 1b. Inflamm Bowel Dis 7:128–132
Weaver CT, Hatton RD, Mangan PR et al (2007) IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol 25:851–852
Kastelein RA, Hunter CA, Cua DJ (2007) Discovery and biology of IL-23 and IL-27: related but functionally distinct regulators of inflammation. Annu Rev Immunol 25:221–242
Ivanov II, McKenzie BS, Zhou L et al (2006) The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126:1121–1133
Langrish CL, Chen Y, Blumenschein WM et al (2005) IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 201:233–240
Zhou L, Lopes JE, Chong MM et al (2008) TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature 453:236–240
Shen W, Durum SK (2010) Synergy of IL-23 and Th17 cytokines: new light on inflammatory bowel disease. Neurochem Res 35:940–946
Bettelli E, Oukka M, Kuchroo VK (2007) TH-17 cells in the circle of immunity and autoimmunity. Nat Immunol 8:345–350
Kinugasa T, Sakaguchi T, Gu X et al (2000) Claudins regulate the intestinal barrier in response to immune mediators. Gastroenterology 118:1001–1011
Rioux JD, Xavier RJ, Taylor KD et al (2007) Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet 39:596–604
Ellson CD, Davidson K, Ferguson GJ et al (2006) Neutrophils from p40phox2/2 mice exhibit severe defects in NADPH oxidase regulation and oxidant-dependent bacterial killing. J Exp Med 203:1927–1937
Ogawa M, Yoshimori T, Suzuki T, Sagara H et al (2004) Escape of intracellular Shigella from autophagy. Science 307:727–731
Lakatos PL, Szamosi T, Szilvasi A et al (2008) ATG16L1 and IL23 receptor (IL23R) genes are associated with disease susceptibility in Hungarian CD patients. Dig Liver Dis 40:867–873
Hampe J, Franke A, Rosenstiel P et al (2007) A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet 39:207–211
Parkes M, Barrett JC, Prescott NJ et al (2007) Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet 39:830–832
Singh SB, Davis AS, Taylor G et al (2006) IRGM induces autophagy to eliminate intracellular mycobacteria. Science 313:1438–1441
Taylor GA (2007) IRG proteins: key mediators of interferon-regulated host resistance to intracellular pathogens. Cell Microbiol 9:1099–1107
Mäkitalo L, Kolho KL, Karikoski R et al (2010) Expression profiles of matrix metalloproteinases and their inhibitors in colonic inflammation related to pediatric inflammatory bowel disease. Scand J Gastroenterol 45:862–871
Salmi M, Andrew DP, Butcher EC et al (1995) Dual binding capacity of mucosal immunoblasts to mucosal and synovial endothelium in humans: dissection of the molecular mechanisms. J Exp Med 181:137–149
Bernstein CN, Sargent M, Rector E (2002) Alteration in expression of beta 2 integrins on lamina propria lymphocytes in ulcerative colitis and Crohn’s disease. Clin Immunol 104:67–72
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bene, L., Falus, A., Baffy, N. et al. Cellular and Molecular Mechanisms in the Two Major Forms of Inflammatory Bowel Disease. Pathol. Oncol. Res. 17, 463–472 (2011). https://doi.org/10.1007/s12253-011-9397-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12253-011-9397-4