
Abstract
Gene silencing due to DNA hypermethylation is a major mechanism for loss of tumor suppressor genes function in colorectal cancer. Activating V600E mutation in BRAF gene has been linked with widespread methylation of CpG islands in sporadic colorectal cancers. The aim of the present study was to evaluate the methylation status of three cancer-related genes, APC2, p14ARF, and ECAD in colorectal carcinogenesis and their association with the mutational status of BRAF and KRAS among Iranian colorectal cancer patients. DNA from 110 unselected series of sporadic colorectal cancer patients was examined for BRAF V600E mutation by PCR-RFLP. Promoter methylation of genes in tumors was determined by methylation specific PCR. The frequency of APC2, E-CAD, and p14 methylation was 92.6%, 40.4% and 16.7%, respectively. But, no V600E mutation was identified in the BRAF gene in any sample. No association was found in cases showing epigenetic APC, ECAD, and p14 abnormality with the clinicopathological parameters under study. The association between KRAS mutations and the so called methylator phenotype was previously reported. Therefore, we also analyzed the association between the hot spot KRAS gene mutations in codons of 12 and 13 with genes’ promoter hypermethylation in a subset of this group of patients. Out of 86 tumors, KRAS was mutated in 24 (28%) of tumors, the majority occurring in codon 12. KRAS mutations were not associated with genes’ methylation in this tumor series. These findings suggest a distinct molecular pathway for methylation of APC2, p14, and ECAD genes from those previously described for colorectal cancers with BRAF or KRAS mutations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC) is the third most common cause of cancer death worldwide. A significant increase in CRC incidence with a predominant distal localization has also been reported in Iran over the last decade [1, 2].
There is increasing evidence suggesting that colorectal cancers develop through different molecular pathways [3]. One of the pathways by which CRC can progress involves transcriptional silencing by hypermethylation of CpG islands, including those present in the promoter regions of tumor suppressor genes, a feature referred as methylator phenotype (CIMP+) [4].
BRAF is a cytoplasmic serine—thereonine kinase that mediates responses to EGF-mediated growth signals through RAS/RAF/MAPK pathway [5]. Activating mutations in the BRAF gene have been identified in 5–15% of sporadic CRCs, more than 80% of which are in the hot spot of exon 15, resulting in V600E substitution [6–8].
The V600E mutation introduces a negative charge in the BRAF that mimics the activating phosphorylation events at nearby threonine and serine residues. Several studies reported a strong association between DNA hypermethylation and BRAF mutation in sporadic CRCs [9–11]. The alteration of KRAS may also contribute to the methylator phenotype in CRC [3]. BRAF and KRAS mutations are important early events in CRC development, and have been strongly correlated with the level of methylation in multiple genes’ promoter in CRC [12]. It has been reported that CRCs with the KRAS mutation were consistent with a methylation frequency between those with a BRAF mutation and those with neither mutation [13].
Aberrant methylation of promoter CpG islands might silence genes that are important for normal cellular homeostasis. Tumor suppressor genes act in several pathways controlling cell proliferation such as cell cycle regulators (p14 and p16), cell adhesion (E-Cadherin), and WNT signaling (APC and APC2) [14, 15].
APC2 is one of the candidate cancer genes (CAN gens) that its epigenetic alterations have been recently identified in CRC by analysis of the DNA hypermethylome in human colorectal cancer [15]. APC2 is a homologue of APC tumor suppressor involved in Wnt signaling pathway. Like APC, APC2 also can inhibit β-catenin signaling and has been suggested to function as a tumor suppressor gene and therefore, important in cancer development [16]. Hypermethylation of APC2 gene has been recently reported in 90% of African-American CRC patients [16, 17]. ECAD is a Ca+-dependent adhesion molecule that mediates intercellular contacts. It plays critical roles in the maintenance of morphogenesis and tissue structure. Loss of ECAD expression is associated with the invasion and metastasis [16, 18]. p14ARF is another tumor suppressor gene that regulates cell cycle and its expression has been shown to enhance stability of p53 through MDM2 suppression. Transcriptional silencing by methylation of CpG islands within the 5′-flanking region and exon 1β of p14 gene has been reported in human CRC [19].
In the present study, we investigated the relationship between APC2, p14, and ECAD promoters’ methylation and BRAF V600E mutation among a group of sporadic CRC patients from south of Iran.
Materials and Methods
Study Population, and Tumor Samples
A total of 110 unselected CRC patients from two referral hospitals in Shiraz, south of Iran were included in this study. Ethics approval for the project was obtained from the Institutional Ethics Committee. Tumor samples were snap frozen immediately after surgical resection and stored at −80°C. All samples were evaluated and subjected to histological diagnosis by expert pathologists and clinical data was obtained. Family history of cancer was analyzed to exclude those pedigrees that met either the Amsterdam I or Amsterdam II criteria. The splenic flexure was used as the anatomical boundary to define proximal and distal CRC.
Methylation-Specific PCR (MS-PCR)
The status of promoter methylation of the APC2, p14, and ECAD genes were determined by MS-PCR. The sequences of primers used for amplification of the promoter regions of genes are listed in Table 1. The MS-PCR reactions were performed as previously described [20]. PCR reactions were “hot started” at 94°C and carried out using 100 ng of bisulfite treated DNA with the following conditions: The PCR reactions were performed in a 50 μL reaction volume containing 25 pmol of each of sense and antisense primers, 0.2 mmol/L dNTPs in 1× PCR buffer provided by the enzyme supplier. The reaction mixture was denatured at 95°C for 5 min, after which 1.5 U Taq polymerase was added; then amplified by 40 cycles, each cycle consisting of 45 s denaturation at 95°C, 45 s annealing at the specified temperatures (Table 1), and 45 s polymerization at 72°C, followed by a single 10-min final extension at 72°C. DNA from peripheral blood lymphocytes was used as negative control in every bisulfite conversion. PCR products were analyzed on 2% agarose gel.
BRAF and KRAS Mutation Analysis
The BRAF genotyping was performed by PCR—RFLP. Genomic DNA from the samples was used as a template in PCR reactions using two BRAF primers encompassing BRAF exon 15 where the V600E substitution is known to take place.
Two nucleotides at the 5′-side of the forward primer were changed (Table 1) to create, in cases of a mutant 1799A nucleotide in the DNA template, an MboII recognition site GAAGA(N8)↓. The wild-type allele with 1799T does not contain the MboII recognition sequence at the corresponding position. MboII digestion of the 97 bp PCR product resulted in 63 bp and 34 bp fragments for the mutant (1799A) allele. Amplified fragments were digested overnight with 10U Mbo II restriction enzyme (MBI Fermentas, Vilnius, Lithuania) at 37°C. The PCR products subjected to enzyme digestion were visualized on 2% agarose gel stained with ethidium bromide. The method was validated by direct sequencing of 50 PCR products.
For the mutational analysis of KRAS, PCR/non-isotopic single strand conformational polymorphism (SSCP) analysis was employed as described by Servomaa et al. [21]. Briefly, codons 12/13 of the KRAS gene were amplified by PCR, as a single fragment, using the primer pair flanking the codons of interest (Table 1). PCR products (2 μl) were denatured for 10 min at 96°C with 2 μl of formamide denaturing dye mixture, cooled on ice, and then applied (4 μl/lane) on 10% nondenaturing polyacrylamide gels. Gels were silver stained and photographed. DNA showing an altered mobility, distinct from that of a normal band in SSCP analysis, was analyzed further by direct sequencing.
Statistical Analysis
Statistical analysis was performed using the SPSS version 11.5 software package (Chicago, IL). Associations between methylation of loci and clinical biological features were evaluated using Chi square and Fisher’s exact test as appropriate.
Results
Distribution of the Selected Characteristics of Cases
In this study, the relationship between promoter methylation of 3 tumor suppressor genes involved in CRC carcinogenesis and BRAF V600E mutation was investigated. One hundred and ten patients entered the study. Selected characteristics of the study population are presented in Table 2. Cases were more likely to be males, smokers and older than 60 years. The incidence of distal tumors was higher than proximal tumors (data not shown). Twenty seven percent (30) of patients had tumors in the proximal colon and 73% (80) in distal parts. No statistically significant differences were found between proximal and distal cancer cases with respect to sex, age, and smoking status. The majority of distal tumors was found to be well/moderately differentiated and was stage II or higher.
APC2, p14 and ECAD Gene Methylation Profiles
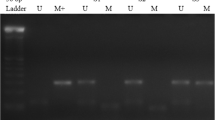
Illustrative examples of MSP reactions for promoter methylation analysis are shown in Fig. 1. The majority of cases showing positive PCR bands for methylated DNA sequences also exhibited them for unmethylated DNA sequences, which were related to tumor heterogeneity or to the occurrence of intermingled normal cells. The epigenetic profile of APC2, p14, and ECAD in CRC is shown in Table 2. We found methylation of APC2 and p14 genes in 104 (95.5%) and 18 (16.4%) of the 110 CRC cases, respectively. The ECAD gene promoter’s methylation was detected in 20 (40.8%) out of 49 CRCs investigated.

Representative examples of MSP reactions for promoter methylation analysis of APC2, ECAD, and p14 genes in primary CRC tumors. The presence of a visible PCR product in those lanes marked U indicates the presence of unmethylated genes; the presence of a product in those lanes marked M indicates the presence of methylated genes. Lane 1 indicates the 100 bp DNA size marker
We analyzed the correlation between the methylation statuses of the genes’ promoter with clinicopathological features. The characteristics of tumors with either one or two (ECAD and p14) methylated genes are summarized in Table 2. When methylation of individual sites was analyzed, no age or sex differences were detected. We also found no significant difference in the methylation status of either gene by tumor location, smoking status, or stage of tumors (Table 2). Coincidental methylation of ECAD and p14 was documented in five out of 49 CRC patients, all being <60 years old (Table 2). A small trend for a higher risk of coincidental alterations of ECAD and p14 was observed in stage I tumors compared with the higher stages.
BRAF and KRAS Mutation Analysis
Samples were analyzed for the presence of a point mutation that frequently occurs in the BRAF oncogene leading to a change of valine to glutamic acid at position 600 of the BRAF protein. The BRAF gene had no detectable mutation in all 110 samples studied using the PCR—RFLP assay. These results suggest that APC2, ECAD, and p14 promoters’ methylation is a frequent event in CRC tumoregenesis and could occur in a different pathway of CRC development from that involving BRAF mutation.
It has been proposed that a subset of CRCs with KRAS mutations may also accumulate promoter methylation in several genes [22]. Therefore, we analyzed the hot spot KRAS gene mutations in codons of 12/13 in a subset of 86 cases of this group of patients (Table 3). We identified oncogenic mutations in 24 out of 86 (28%) tumors, the majority occurring in codon 12 and a few in codon 13. A significant difference in the distribution of KRAS mutation was observed between patients with proximal colon tumor and those with distal CRC, as verified by the Fisher’s exact test (p = 0.002). KRAS mutations were not associated with genes’ methylation in this tumor series (Table 4). Together, these results suggest for the existence of alternate CIMP pathways of CRC development to those involving BRAF and KRAS mutations.
Discussion
Our data indicates extensive methylation of the APC2 and ECAD genes in CRC patients. We observed a high frequency of 95.5% methylation in the APC2 promoter. Gene’s promoter methylation was not specifically associated with any of the clinicopathological parameters considered in this study. In a recent population-based study, a similar frequency of 96% methylation of the APC2 gene in African- Americans and 94% in Iranian sporadic CRC patients were reported [23]. APC2 is a homologue of APC1 tumor suppressor involved in Wnt signaling pathway. In our previous study in this group of patients, the frequency of APC1 methylation was 11.8% (unpublished). Therefore, it seems that the APC2 hypermethylation is a more frequent event than the APC1 hypermethylation in colorectal carcinogenesis.
ECAD was hypermethylated in 40.8% of primary CRCs, a finding consistent with previous reports. Miranda et al. reported a frequency of 44% for ECAD methylation [24]. In another study, half of all CRC samples were found to be hypermethylated in the ECAD promoter, although the sample size was relatively small [25]. Mutations of the ECAD gene have been only rarely found in most types of sporadic cancers [26]. It seems that promoter methylation is the major oncogenic event for the ECAD gene inactivation in CRC.
In the previous studies, hypermethylation of the promoter region in the p14ARF gene was reported in 18–33% of human colon cancers [19, 27]. We detected p14 methylation in 16% of the cases. MSI has been classified into two categories, high-level MSI (MSI-H) and low-level MSI (MSI-L). In a recent study of 234 CRCs, promoter methylation of p14 was significantly associated with MSI-low (MSI-L) CRC and with KRAS mutation [13]. The authors concluded that promoter methylation of p14 could be an alteration leading to a CRC with MSI-L. The CRC series investigated in this study have also been characterized previously for MSI + and the methylation status of hMLH1, and p16 promoters [20, 28]. Therefore, this allowed us to examine the association between KRAS mutation with hypermethylation and MSI status in the present study. MSI analysis was performed using the reference panel of five markers recommended by the “Bethesda guidelines” (D2S123, D5S346, D17S250, BAT25, and BAT26). Tumors were classified as MSI positive if two or more markers showed instability [28]. We found MSI+ tumors in 23.8% of this CRC series and KRAS mutations in 28% of tumors (Table 3). We did not find an association between promoter methylation of the p14 gene and MSI tumors with KRAS mutations in these CRC tumors (Table 4). BRAF mutation was also reported to positively associate with MSI + status and hMLH1 methylation [11, 29]. We verified the relationship between the BRAF gene mutation and the MSI and the hypermethylation of hMLH1 gene in this group of patients. As described above, while there was no BRAF mutation in 110 CRCs investigated, 23.8% of the patients were MSI + and 13.2% had methylated hMLH1 promoter.
We also analyzed the differences in the methylation status of these three tumor-related genes in CRC patients according to their clinicopathological features. We found no significant differences of methylation status of APC2, p14, and E-CAD genes between these subgroups (Table 2). The frequency of coincidental methylation of ECAD and p14 genes was higher in younger patients (<60 years) than that in the old ones. A small trend for a higher risk of coincidental methylation of p14 and ECAD was observed in stage I tumors (Table 2).
In our previous study on this group of cases, we identified 13.2% methylation of hMLH1, 2.6% methylation of hMSH2, and 19.9% methylation of p16 genes [20]. About half of the promoters of human genes have CpG islands, and are susceptible to hypermethylation. But, it is unclear why certain genes are more frequently affected than others.
There is not yet a consensus on what markers and criteria should be used for classification of CIMP in CRC. A panel of five markers including RUNX3, CACNA1G, IGF2, NEROG1, and SOCS1 were recently proposed for defining CIMP+ tumors [10, 30]. Using above markers, the frequency of CIMP+ was estimated at approximately 15–18% of CRC patients.
It has been previously reported that BRAF and KRAS mutations are associated with different levels of DNA hypermethylation [12]. CRCs with BRAF mutations are associated with a high-level of promoter methylation in multiple loci, but tumors with KRAS mutations are associated with a low-level of promoter methylation. CRCs with neither KRAS nor BRAF mutations have very little methylation. BRAF somatic mutations were reported in ~15% of sporadic CRCs [7, 8]. We found no BRAF mutations in this study confirming recent findings of Brim et al. [31] reporting the lowest mutation rate of this gene among Iranian (2%) in comparison to Omanis (19%), and African American (10%) patients. Brim et al. used direct sequencing method and we employed PCR-RFLP to analyze the presence of BRAF mutation. The difference is unlikely to be due to a failure of our mutation detection methodology because we also verified PCR-RFLP results by direct sequencing in a subset of our samples (see “Materials and Methods” section). We assume that the genetic or environmental factors may underlie the lack of BRAF mutation among Iranian CRC patients.
We detected a substantial promoter methylation in genes investigated in 110 CRC specimens. Our results showed the lack of association of the methylation status with either BRAF or KRAS mutations in this group of CRC patients. Despite the size limitations, our preliminary data suggest possibly a distinct molecular pathway for methylation of APC2, p14, and E-CAD genes from those previously described for CRCs with BRAF or KRAS mutations. Further studies will be necessary to substantiate this issue.
References
Malekzadeh R, Bishehsari F, Mahdavinia M, Ansari R (2009) Epidemiology and molecular genetics of colorectal cancer in Iran: a review. Arch Iran Med 12:161–169
Fazeli MS, Adel MG, Lebaschi AH (2007) Colorectal carcinoma: a retrospective, descriptive study of age, gender, subsite, stage, and differentiation in Iran from 1995 to 2001 as observed in Tehran University. Dis Colon Rectum 50:990–995
Jass JR (2007) Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology 50:113e30
Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP (1999) CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA 96:8681–8686
Marais R, Marshall CJ (1996) Control of the ERK MAP kinase cascade by Ras and Raf. Cancer Surv 27:101–125
Li WQ, Kawakami K, Ruszkiewicz A, Bennett G, Moore J, Iacopetta B (2006) BRAF mutations are associated with distinctive clinical, pathological and molecular features of colorectal cancer independently of microsatellite instability status. Mol Cancer 5:2
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S et al (2002) Mutations of the BRAF gene in human cancer. Nature 417:949–954
Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE (2002) Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature 418:934
Suehiro Y, Wong CW, Chirieac LR, Kondo Y, Shen L, Webb CR et al (2008) Epigenetic-genetic interactions in the APC/WNT, RAS/RAF, and P53 pathways in colorectal carcinoma. Clin Cancer Res 14:2560–2569
Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA et al (2006) CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet 38:787–793
English DR, Young JP, Simpson JA, Jenkins MA, Southey MC, Walsh MD et al (2008) Ethnicity and risk for colorectal cancers showing somatic BRAF V600E mutation or CpG island methylator phenotype. Cancer Epidemiol Biomark Prev 17:1774–1780
Nagasaka T, Sasamoto H, Notohara K, Cullings HM, Takeda M, Kimura K et al (2004) Colorectal cancer with mutation in BRAF, KRAS, and wild-type with respect to both oncogenes showing different patterns of DNA methylation. J Clin Oncol 22:4584–4594
Kominami K, Nagasaka T, Cullings HM, Hoshizima N, Sasamoto H, Young J et al (2009) Methylation in p14(ARF) is frequently observed in colorectal cancer with low-level microsatellite instability. J Int Med Res 37:1038–1045
Auerkari EI (2006) Methylation of tumor suppressor genes p16 (INK4a), p27(Kip1) and E-cadherin in carcinogenesis. Oral Oncol 42:5–13
Schuebel KE, Chen W, Cope L, Glöckner SC, Suzuki H, Yi JM et al (2007) Comparing the DNA hypermethylome with gene mutations in human colorectal cancer. PLoS Genet 3:1709–1723
Kumar K, Brim H, Giardiello F, Smoot DT, Nouraie M, Lee EL et al (2009) Distinct BRAF (V600E) and KRAS mutations in high microsatellite instability sporadic colorectal cancer in African Americans. Clin Cancer Res 15:1155–1161
Garinis GA, Menounos PG, Spanakis NE, Papadopoulos K, Karavitis G, Parassi I et al (2002) Hypermethylation-associated transcriptional silencing of E-cadherin in primary sporadic colorectal carcinomas. J Pathol 198:442–449
Takeichi M (1995) Morphogenetic roles of classic cadherins. Curr Opin Cell Biol 7:619–627
Zheng S, Chen P, McMillan A, Lafuente A, Lafuente MJ, Ballesta A et al (2000) Correlations of partial and extensive methylation at the p14(ARF) locus with reduced mRNA expression in colorectal cancer cell lines and clinicopathological features in primary tumors. Carcinogenesis 21:2057–2064
Mokarram P, Naghibalhossaini F, Saberi Firoozi M, Hosseini SV, Izadpanah A, Salahi H et al (2008) Methylenetetrahydrofolate reductase C677T genotype affects promoter methylation of tumor-specific genes in sporadic colorectal cancer through an interaction with folate/vitamin B12 status. World J Gastroenterol 14:3662–3671
Servomaa K, Kiuru A, Kosma VM, Hirvikoski P, Rytömaa T (2000) p53 and K-ras gene mutations in carcinoma of the rectum among Finnish women. Mol Pathol 53:24–30
Gazin C, Wajapeyee N, Gobeil S, Virbasius CM, Green MR (2007) An elaborate pathway required for Ras-mediated epigenetic silencing. Nature 449:1073–1077
Mokarram P, Kumar K, Brim H, Naghibalhossaini F, Saberi-firoozi M, Nouraie M et al (2009) Distinct high-profile methylated genes in colorectal cancer. PLoS ONE 4:e7012
Miranda E, Destro A, Malesci A, Balladore E, Bianchi P, Baryshnikova E et al (2006) Genetic and epigenetic changes in primary metastatic and nonmetastatic colorectal cancer. Br J Cancer 95:1101–1107
Wheeler JM, Kim HC, Efstathiou JA, Ilyas M, Mortensen NJ, Bodmer WF (2001) Hypermethylation of the promoter region of the E-cadherin gene (CDH1) in sporadic and ulcerative colitis associated colorectal cancer. Gut 48:367–371
Strathdee G (2002) Epigenetic versus genetic alterations in the inactivation of E-cadherin. Semin Cancer Biol 12:373–379
van Engeland M, Weijenberg MP, Roemen GM, Brink M, de Bruïne AP, Goldbohm RA et al (2003) Effects of dietary folate and alcohol intake on promoter methylation in sporadic colorectal cancer: the Netherlands cohort study on diet and cancer. Cancer Res 63:3133–3137
Naghibalhossaini F, Mokarram P, Khalili I, Vasei M, Hosseini SV, Ashktorab H et al (2010) MTHFR C677T and A1298C variant genotypes and the risk of microsatellite instability among Iranian colorectal cancer patients. Cancer Genet Cytogenet 197:142–151
Yuen ST, Davies H, Chan TL, Ho JW, Bignell GR, Cox C et al (2002) Similarity of the phenotypic patterns associated with BRAF and KRAS mutations in colorectal neoplasia. Cancer Res 62:6451–6455
Ogino S, Kawasaki T, Kirkner GJ, Kraft P, Loda M, Fuchs CS (2007) Evaluation of markers for CpG island methylator phenotype (CIMP) in colorectal cancer by a large population-based sample. J Mol Diagn 9:305–314
Brim H, Mokarram P, Naghibalhossaini F, Saberi-Firoozi M, Al-Mandhari M, Al-Mawaly K et al (2008) Impact of BRAF, MLH1 on the incidence of microsatellite instability high colorectal cancer in populations based study. Mol Cancer 7:68
Acknowledgments
We thank Dr. Mahmood Vessal of Shiraz University of Medical Sciences for carefully reading the manuscript. This study was part of the dissertation of Hamideh Mahmoodzadeh Hosseinini, submitted to Shiraz University of Medical Sciences in partial fulfillment of the requirements for the MSc in biochemistry. This work was supported by a grant from the Vice Chancellor for Research, Shiraz University of Medical Sciences.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Naghibalhossaini, F., Hosseini, H.M., Mokarram, P. et al. High Frequency of Genes’ Promoter Methylation, but Lack of BRAF V600E Mutation among Iranian Colorectal Cancer Patients. Pathol. Oncol. Res. 17, 819–825 (2011). https://doi.org/10.1007/s12253-011-9388-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12253-011-9388-5