
Abstract
Heterologously expressed and purified azoreductase enzyme from facultative Klebsiella pneumoniae was used to degrade sulphonated azo dye. Methyl orange (MO) was used as the model dye to study the azo dye decolorization potential of the purified enzyme at different conditions. The enzyme had maximum activity at 40 °C and pH 8.0. The enzyme was observed to be thermo-stable as some enzyme activity was retained even at 80 °C. The apparent kinetic parameters, i.e., appKm and appVmax, for azoreductase using MO as a substrate were found to be 17.18 μM and 0.08/min, respectively. The purified enzyme was able to decolorize approximately 83% of MO (20 μM) within 10 min in the presence of NADH. Thus, efficient decolorization of MO was observed by the purified enzyme. The recombinant enzyme was purified approximately 18-fold with 46% yield at the end of four steps of the purification process. Enzyme was present in a tetrameric structure as confirmed by the volume at which protein was eluted in gel filtration chromatography, and the monomeric molecular mass of enzyme was found to be 23 kDa on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The dye degradation efficiency of azoreductase cloned from Klebsiella pneumoniae and purified from recombinant Escherichia coli was observed to be much higher as compared with the efficiencies of the reported azoreductases from other bacterial strains. In the present study, we report the purification and characterization of the azoreductase cloned from Klebsiella pneumoniae and expressed in Escherichia coli.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The use of azo dyes has been reported in different industries such as paper, food, cosmetics, and pharmaceuticals with the largest consumer being the textile industries (Fontenot et al. 2003; Padamavathy et al. 2003). Azo dyes are considered as one of the major environmental pollutants due to their xenobiotic nature (Carliell et al. 1995; Chung and Stevens 1993). Most of these dyes are toxic, and their biotransformation products are known to be carcinogenic and mutagenic (Pinheiro et al. 2004). Microbial degradation is considered as one of the ecofriendly and cost-effective methods to treat wastewater containing azo dye contamination (Sudha et al. 2014).
Klebsiella pneumoniae is reported to be present in toxic dye-contaminated soil and wastewater. The ability of Klebsiella pneumoniae to degrade these toxic azo dyes is also reported in a few studies (Cui et al. 2014; Franciscon et al. 2009). In one of the previously reported studies, Wong and Yuen isolated Klebsiella pneumoniae from dye-contaminated sludge which showed the ability to decolorize Methyl red azo dye (Wong and Yuen 1996). Recently, Li and co-workers reported the biodegradation of malachite green by Klebsiella pneumoniae strain WA-1 (Li et al. 2019). The decolorization and mineralization of four different azo dyes by Klebsiella sp. strain VN-31 was also reported (Franciscon et al. 2009). Klebsiella sp. Y3 was reported in recent years to decolorize different azo dyes (Cui et al. 2014). The decolorization of two different azo dyes using two different Klebsiella strains was also reported by Zabłocka-Godlewska and co-workers (Zabłocka-Godlewska et al. 2014).
In general, bacterial systems have developed specific enzymes for azo dye reduction under specific environmental conditions. Azoreductases are the enzymes which catalyze the reduction of azo dyes into their corresponding amines under anaerobic condition as the first step of the azo dye biodegradation process (Stolz 2001). In the past few decades, many attempts have been made to isolate and purify azoreductase enzyme to understand the structure and function of azoreductases from different bacterial strains (Chen et al. 2004; Ooi et al. 2007). These studies have helped in elucidating their specific and broad range substrate activities. Thus, it is essential to purify and characterize azo dye degrading enzymes, i.e., azoreductases, from different microbes involved in the azo dye degradation. This may result in obtaining better insights into the decolorization processes.
Isolation and characterization of several azoreductase enzymes are reported from different species of bacteria over the last few decades. Zimmermann and co-workers isolated and purified the azoreductase from a Pseudomonas strain KF46 for the first time which decolorized Orange II efficiently (Zimmermann et al. 1982). An oxygen-insensitive intracellular azoreductase of 27 kDa which could decolorize Methyl red was purified and characterized from Escherichia coli CD-2 by Cui and co-workers (Cui et al. 2012). Similarly, Moutaouakkil and co-workers purified monomeric azoreductase of molecular mass 28 kDa from Enterobacter agglomerans which could decolorize Methyl red and related azo dyes (Moutaouakkil et al. 2003). Flavin-dependent azoreductase was successfully purified by Punj and John from Enterococcus faecalis (Punj and John 2009). Bin and co-workers expressed azoreductase from Rhodobacter sphaeroides AS1.1737 into Escherichia coli and purified azoreductase with molecular mass of 18.7 kDa (Bin et al. 2004). The thermo-stable azoreductase was purified by Misal and co-workers from Bacillus badius, and the purified azoreductase showed 43 kDa molecular mass on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (Misal et al. 2011). In a previous study, the isolated facultative Klebsiella pneumoniae decolorized Methyl Orange (MO, 100 mg/L) in 3 h when media was supplemented with NADH (Dixit and Garg 2018). The dye decolorization rate was observed to be much faster in comparison with other sub-strains of Klebsiella sp. reported in the literature. It is known that a facultative Klebsiella sp. strain VN-31 decolorized 100 mg/L of four azo dyes, RY107, RR 198, RB 5, and DB 71 in 72, 96, 120, and 168 h, respectively, under microaerophilic/aerated sequential process (Franciscon et al. 2009). Similarly, it is also reported that under anaerobic condition, Klebsiella sp. strain Y3 decolorized four different azo dyes in 48 h (Cui et al. 2014). Zabłocka-Godlewska and co-workers also reported the decolorization of two azo dyes using two different Klebsiella strains (Zabłocka-Godlewska et al. 2014). In a recent study, Li and co-workers have reported the biodegradation of malachite green by Klebsiella pneumoniae strain WA-1 (Li et al. 2019).
The purification of a previously cloned and heterologously expressed azoreductase from Klebsiella pneumoniae into Escherichia coli (Dixit and Garg 2018) was performed using different purification techniques and is reported in the present study. The purified recombinant azoreductase was characterized, and its apparent kinetic properties were also evaluated. The azoreductase encoding gene was initially amplified from Klebsiella pneumoniae. The azoreductase gene was amplified through polymerase chain reaction and cloned in pGEM-T vector and further expressed in Escherichia coli (Dixit and Garg 2018).
Materials and methods
Materials
Q Sepharose HP 5 mL and Mono Q 10/100 GL columns used in this study for anion exchange chromatography were procured from GE Healthcare Life Sciences, India. Superdex 75 10/300 GL column was used for gel filtration chromatography and procured from Sigma-Aldrich. The standard protein ladder was procured from Genei, Bangalore, India. Ammonium sulfate and isopropyl-β-D-thiogalactopyranoside (IPTG) were purchased from SRL Pvt. Ltd., India. The azo dye, MO, used in this study was procured from SD Fine CHEM Limited, Mumbai, India. NADH was purchased from Loba Chemie, India. All other reagents and chemicals used for the experiments were of analytical grade.
Microorganism and growth conditions
The pET-28b-azoR gene inserted recombinant Escherichia coli BL21 (DE3) bacterial cell culture (Dixit and Garg 2018) was grown overnight at 37 °C in 10 mL LB broth supplemented with kanamycin (50 μg/mL). Ten milliliters of this culture was used to inoculate 2 L LB broth at 37 °C. The culture was supplemented with antibiotic kanamycin at a concentration of 50 μg/mL. The culture was allowed to grow further at shaking condition of 120 rpm until OD reached to 0.66 at 600 nm. Subsequently, the culture was supplemented with 1 mM IPTG to initiate azoreductase gene expression. The culture with IPTG was further grown for 16 h at 20 °C.
Preparation of crude extract
Two liters of induced culture was centrifuged (at approximately 6583 g, 4 °C) for 15 min to harvest the recombinant cells. The cells were washed with low salt buffer (25 mM Tris-Cl, 100 mM NaCl, pH 8.0) and resuspended in 50 mL lysis buffer containing 1 mM phenylmethylsulfonyl fluoride (PMSF, a protease inhibitor). Cells were disrupted on ice by sonication for 1 h (15 s on and 30 s off cycles, 70% output) using Sonics Vibra Cell Sonicator. The centrifugation of sonicated cells (at approximately 67,779 g, 4 °C) for 30 min was done using Thermo Fisher Scientific Sorvall Lynx 6000 centrifuge to separate pellet (cell debris) from supernatant. The pellet was stored at 4 °C. The bacterial crude extract containing soluble protein fraction was obtained as supernatant. The separated crude extract was used for further purification steps.
Purification steps
Enzyme purification was performed using the following steps at 4 °C.
Ammonium sulfate precipitation
The bacterial crude extract was precipitated with solid ammonium sulfate (NH4)2SO4 at 20% (w/v) saturation by stirring at 4 °C for 20 min on a magnetic stirrer (Moutaouakkil et al. 2003). Centrifugation (at approximately 24,400 g, 4 °C) of the stirred solution was done for 20 min to separate the precipitated protein as pellet which was stored at 4 °C. Requisite amount of ammonium sulfate salt was further added in the supernatant to bring the solution to 40% (w/v) saturation. The 40% saturated solution was stirred for 20 min followed by centrifugation (at approximately 24,400 g, 4 °C) for 20 min to get pellet and supernatant. Similar procedure of salt addition and centrifugation was repeated to obtain 60% and 80% saturated solutions. The stored pellets, obtained from different saturated solutions, were dissolved in 2 mL of low salt buffer. The dissolved pellets were analyzed on SDS-PAGE. The pellet giving the maximum concentration of the desired protein was used further in dialysis to remove excess salts. The protein solution was dialyzed (dialysis membrane, 14 kDa, Sigma, India) initially in low salt buffer (2 L, 4 h) and finally in fresh low salt buffer (2 L, overnight). Two stages of ion-exchange chromatography followed by one stage of gel filtration chromatography were further used to purify the recombinant protein, azoreductase (Chen et al. 2004).
Q Sepharose ion-exchange chromatography
The protein solution obtained after dialysis was diluted with low salt buffer and applied to Q Sepharose HP 5 mL column. The column was equilibrated and washed with low salt buffer (5 mL/min), and the diluted sample was then passed through the column (1 mL/min). The adsorbed proteins were eluted using a linear concentration gradient of sodium chloride, NaCl (75 mM to 1 M). Two milliliters of sample fractions was collected, and the fractions with azoreductase activity were selected for further analysis. These selected fractions were further dialyzed at 4 °C in 2 L low salt buffer for 4 h and used in the next purification step.
Mono Q ion-exchange chromatography
Equilibration of Mono Q column was done with low salt buffer followed by extensive washing (5 mL/min) for 2 h with the buffer. The previously dialyzed protein fraction was diluted with low salt buffer and was passed through (2 mL/min) pre-equilibrated Mono Q 10/100 GL column. NaCl gradient of 75 mM to 1 M was applied linearly to elute the protein. Two milliliters of sample fractions of the eluted proteins was collected, and the active fractions were selected. The selected fractions were concentrated to about 2 mL using centricon cutter (10 kDa cut off, Millipore, India) using a refrigerated centrifuge.
Gel filtration chromatography
Equilibration of Superdex 75 10/300 GL column was done with low salt buffer solution. The concentrated protein solution obtained after Mono Q purification was passed through the pre-equilibrated Superdex column with gel filtration buffer (25 mM Tris-Cl, 100 mM NaCl, 5% glycerol) at 0.4 mL/min. Two milliliters of sample fractions was collected. The fractions with azoreductase activity were selected and stored at − 20 °C until further use.
Azoreductase activity assay
The enzyme activity was measured as per the reported method of Zimmermann and co-workers (Zimmermann et al. 1982). The azoreductase activity was monitored by the decrease in absorbance of azo dye at its characteristic wavelength using Thermo Scientific Multiskan Spectrum spectrophotometer at 25 °C. The standard reaction mixture (2 mL) contained Tris-Cl buffer (25 mM, pH 8.0), MO (50 μM), NADH (0.2 mM), and a suitable amount of azoreductase. The reaction mixture was pre-incubated at 30 °C without NADH for 10 min. NADH was subsequently added to the reaction mixture to initiate the reaction. MO decolorization was measured using absorbance values at 467 nm wavelength. One unit of azoreductase activity (U, μM/min) was defined as the amount of enzyme required to reduce 1 μM azo dye per minute as also defined earlier by Misal and co-workers (Misal et al. 2011). All assays were done in triplicate.
Protein concentration estimation
The selected purified protein concentration was estimated using BCA procedure as per the manufacturer’s instructions where bovine serum albumin (BSA) was used as a standard. The bicinchoninic acid assay (BCA) kit (G-Biosciences, India) was used to conduct the protein estimation experiments (Chen et al. 2004; Walker 2009).
Analytical gel electrophoresis
SDS–PAGE of the concentrated purified protein was done as per the reported method (Laemmli 1970). SDS-PAGE was done on a vertical gel unit (Tarsons® vertical SDS-PAGE unit). Twelve percent polyacrylamide and 0.1% SDS was used according to the molecular mass of the protein. SDS-PAGE was run initially at 50 V for 30 min followed by 150 V for nearly 2 h. After complete run, gels were stained with Coomassie Brilliant Blue R-250 dye in a mixture of water, methanol, and acetic acid in a volumetric ratio of 1/2, 2/5, and 1/10, respectively, for 0.5 h on a rocker. Further destaining was done in a mixture of water, methanol, and acetic acid in a volumetric ratio of 1/2, 2/5, and 1/10, respectively, until the protein bands had become visible and the background dye was removed. The relative mobility of the target protein as compared with the protein molecular mass markers (Genei, Bangalore, India) was used to calculate its molecular mass.
Optimization of different parameters for purified azoreductase
A wide range of buffer systems, acetate buffer (pH 5), phosphate buffer (pH 6-7), and Tris-Cl buffer (pH 8-9) were used to measure the optimum pH for azoreductase activity. The effect of pH on azoreductase activity was determined using ELISA plate reader. The reaction mixture was incubated at different pH at 37 °C for 30 min. The azoreductase activity was measured after incubation of 30 min in 25 mM Tris-Cl buffer at pH 8.0 to determine the optimal temperature. The activity values were measured over a temperature range of 20–60 °C. ELISA plate reader spectrophotometer was used to measure the activity of different samples. All experiments were done in triplicate.
Decolorization of MO by purified azoreductase
To determine activity of the purified azoreductase, MO decolorization studies at different concentrations (20–60 μM) were performed in 2 mL reaction mixture. In 2 mL reaction mixture, 25 mM Tris-Cl buffer and suitable amounts of dye and azoreductase at pH 8.0 were mixed. To initiate the reaction, 0.2 mM NADH was subsequently added, and the reaction mixture was incubated at 37 °C for 30 min. The rate of MO decolorization by azoreductase was estimated using the change in absorbance at 467 nm for different samples (collected at different time intervals) using spectrophotometer.
Kinetic studies of purified azoreductase
To determine the apparent kinetic parameters (apparent Michaelis–Menten constant (appKm) and apparent maximum velocity (appVmax)) of sulphonated azo dye decolorization using azoreductase enzyme, the concentration of one of the substrates was varied at a time keeping the other substrate concentration constant. MO concentration was varied from 20 to 60 μM, while the concentration of NADH was kept constant. appKm and appVmax were estimated using the collected data and Lineweaver–Burk plot (Lineweaver and Burk 1934).
Results and discussion
Purification of azoreductase
In a previous study, azoreductase gene from Klebsiella pneumoniae was cloned in pET-28 b (+) vector. The previously cloned gene was expressed in Escherichia coli BL21 using 1 mM IPTG and expression was confirmed on SDS-PAGE. The recombinant Escherichia coli BL21 cells were dissolved in lysis buffer and sonicated to get the crude extract. The obtained crude extract was used to get the purified azoreductase using a four-step purification process. The heterologously expressed azoreductase was purified using ammonium sulfate precipitation followed by two-step ion-exchange column chromatography (Q Sepharose, Mono Q) and one-step gel filtration chromatography. Dialysis was used after ammonium sulfate precipitation and in between the three chromatography steps to remove the salts. The total protein obtained from the crude extract was 1.769 mg with a total activity of 8.61 × 10+3 U. Table 1 summarizes the purification level obtained at each step. The first step of the protein purification was ammonium sulfate (NH4)2SO4 precipitation which eliminated other proteins and was followed by dialysis. At the end of this step, 1.015 mg protein was obtained with a total activity of 7.21 × 10+3 U. The second and third steps involved ion-exchange column chromatography (Q Sepharose and Mono Q, respectively), and the fourth step was gel filtration chromatography (GFC). Finally, the concentrated enzyme solution was passed through a Superdex 75 column, and the active fraction of the azoreductase was eluted at 9 mL volume (supplement Fig. S1). The purified recombinant azoreductase was observed to be tetrameric with molecular mass of 92 kDa (based on the low molecular mass gel filtration calibration kit for Superdex 75 10/300 GL column). The final total protein after GFC purification was observed to be 0.045 mg with a total activity of 3.96 × 10+3 U. After all the four purification steps, the yield of azoreductase was 46% with 18-fold purification factor and 88 × 10+3 U/mg specific activity. In reported studies, yield of the purified azoreductase from Enterococcus faecalis and Staphylococcus aureus ATCC 25923 were only 21% and 20%, respectively (Chen et al. 2005; Chen et al. 2004). The purified recombinant azoreductase showed 23 kDa molecular mass on SDS-PAGE and was observed to be present in tetrameric form. NADH was required as an electron donor by the purified azoreductase for azo bond reduction. Similarly, azoreductase isolated from Enterococcus faecalis also had 23 kDa molecular mass with NADH as a preferred electron donor (Chen et al. 2004). Although azoreductase from Klebsiella pneumoniae and the reported azoreductase from Enterococcus faecalis had similar molecular masses, they did not had any amino acid sequence similarity.
Molecular mass determination
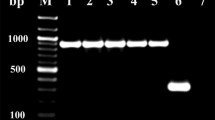
The SDS–PAGE analyses of different protein fractions obtained after each purification step were done, and the results are shown in Fig. 1. A single band corresponding to the purified azoreductase of Klebsiella pneumoniae was observed on SDS-PAGE as shown in Fig. 1 with 23 kDa molecular mass. Azoreductases from several other bacterial strains, such as Bacillus sp. strain B29 (Ooi et al. 2009), Enterococcus faecalis strain ATCC 19433 (Chen et al. 2004), Pigmentiphaga kullae K24 (Chen et al. 2010), and Pseudomonas aeruginosa strain PAO1 (Wang et al. 2007), have shown approximately similar molecular masses. Azoreductases from Klebsiella pneumoniae are not yet reported in the open literature.

SDS-PAGE of Klebsiella pneumoniae azoreductase purified from Escherichia coli BL21. Lane M, protein molecular marker; lane 1, crude extract; lane 2, 40% ammonium sulfate precipitation (ASP); lane 3, Q Sepharose; lane 4, Mono Q; and lane 5, purified azoreductase
Optimal parameters for the purified azoreductase
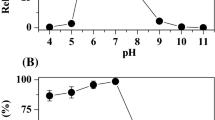
In this study, the enzyme assay for the purified azoreductase was performed using MO as a model substrate in the presence of NADH. The azoreductase activity was optimized for different parameters like pH, temperature, and substrate. Different pH buffers with pH range of 5.0 to 9.0 were used to determine the effect of pH on the purified azoreductase activity. The purified azoreductase had bell-shaped curve over the selected pH range. The maximum activity was observed at pH 8.0 as shown in Fig. 2a, which is similar to the optimal pH activity of azoreductase from Rhodobacter sphaeroides AS1.1737 (Bin et al. 2004). It was observed that there was a drastic decrease in the azoreductase activity both at pH below and above 8.0. It was noted that purified azoreductase from Klebsiella pneumoniae was stable at pH 8.0 as more than 80% activity was observed after incubation of the reaction mixture for 30 min at 40 °C.

Characterization of purified azoreductase. (a) pH effect on the purified azoreductase activity, (b) temperature effect on the purified azoreductase activity, and (c) double reciprocal plot for varying concentration of MO
Different temperatures in the range 30 to 80 °C were used to study the effect of temperature on the purified azoreductase activity at constant pH 8.0. The purified azoreductase had its maximum activity at temperature 40 °C which is similar to the reported optimal temperature for azoreductases from Rhodobacter sphaeroides AS1.1737 and Pigmentiphaga kullae K24, etc. (Bin et al. 2004; Chen et al. 2010). It was noted that there was a decrease in the activity of the purified azoreductase at temperatures below and above 40 °C as shown in Fig. 2b. However, the purified azoreductase was thermo-stable as it retained some activity even at temperature of 80 °C. The purified azoreductase nearly lost its activity at 20 °C. Thus, the optimum temperature and pH were observed to be 40 °C and 8.0 for azoreductase activity, respectively. These were observed to be in agreement with the reported values for azoreductases from Rhodobacter sphaeroides AS1.1737 and Pigmentiphaga kullae K24 (Bin et al. 2004; Chen et al. 2010).
MO decolorization by the purified azoreductase
In the present study, the decolorization efficiency (Cui et al. 2012) of the purified azoreductase to decolorize MO was studied. MO decolorization study by the purified azoreductase at different initial concentrations of the dye and optimal pH and temperature values was also carried out (supplement Fig. S2). Almost 68% dye (20 μM initial dye concentration) was decolorized in 6 min. After incubation for 10 min, 83% decolorization was observed. For 60 μM concentration of MO, decolorization was only 60% within 6 min and was approximately 80% after 10 min of incubation. These results indicate that the purified azoreductase efficiently degraded MO at its optimum operating conditions.
appK m and appV max for the purified azoreductase
Kinetic studies were also carried out for estimating azoreductase activity using MO as a substrate at different concentrations while keeping NADH concentration constant. The values of apparent kinetic parameters, appKm and appVmax, for purified azoreductase, were calculated from the double reciprocal plot (Lineweaver–Burk plot) as shown in Fig. 2c. The concentration of MO was varied keeping the concentration of NADH constant. The appKm and appVmax values for purified azoreductase using MO as the substrate were calculated to be 17.18 μM and 0.08/min, respectively. The lower appKm value showed high affinity of the enzyme for MO. Thus, on the basis of observed results in the present study, it was established that azoreductase from Klebsiella pneumoniae could be utilized to rapidly degrade sulphonated azo dyes present in wastewaters.
Conclusions
Heterologously expressed and purified azoreductase enzyme from facultative Klebsiella pneumoniae was efficiently used to decolorize sulphonated azo dye. The purified enzyme decolorized different concentrations of MO efficiently at the optimal pH 8 and temperature of 40 °C in the presence of NADH. The observed results indicate the potential use of the recombinant Escherichia coli to decolorize dye-contaminated wastewaters at different source points in the near future.
Data availability
Experimental data may be obtained on request by email from Shweta Dixit. The wild-type Klebsiella pneumoniae is available from the Microbial Culture Collection (MCC), Pune, India, under accession number MCC3212.
References
Bin Y, Jiti Z, Jing W, Cuihong D, Hongman H, Zhiyong S, Yongming B (2004) Expression and characteristics of the gene encoding azoreductase from Rhodobacter sphaeroides AS1.1737. FEMS Microbiol Lett 236:129–136
Carliell CM, Barclay SJ, Naidoo N, Buckley CA, Mulholland DA, Senior E (1995) Microbial decolourisation of a reactive azo dye under anaerobic conditions. Water SA 21:61–69
Chen H, Feng J, Kweon O, Xu H, Cerniglia CE (2010) Identification and molecular characterization of a novel flavin-free NADPH preferred azoreductase encoded by azoB in Pigmentiphaga kullae K24. BMC Biochem 11:1–10
Chen H, Hopper SL, Cerniglia CE (2005) Biochemical and molecular characterization of an azoreductase from Staphylococcus aureus, a tetrameric NADPH-dependent flavoprotein. Microbiology (Reading, England) 151:1433–1441
Chen H, Wang RF, Cerniglia CE (2004) Molecular cloning, overexpression, purification, and characterization of an aerobic FMN-dependent azoreductase from Enterococcus faecalis. Protein Expr Purif 34:302–310
Chung KT, Stevens SE (1993) Degradation azo dyes by environmental microorganisms and helminths. Environ Toxicol Chem 12:2121–2132
Cui D, Li G, Zhao D, Gu X, Wang C, Zhao M (2012) Purification and characterization of an azoreductase from Escherichia coli CD-2 possessing quinone reductase activity. Process Biochem 47:544–549
Cui D, Li G, Zhao M, Han S (2014) Decolourization of azo dyes by a newly isolated Klebsiella sp. strain Y3, and effects of various factors on biodegradation. Biotechnol Biotechnol Equip 28:478–486
Dixit S, Garg S (2018) Development of an efficient recombinant bacterium and its application in the degradation of environmentally hazardous azo dyes. Int J Environ Sci Technol. https://doi.org/10.1007/s13762-13018-12054-13767
Fontenot EJ, Lee YH, Matthews RD, Zhu G, Pavlostathis SG (2003) Reductive decolorization of a textile reactive dyebath under methanogenic conditions. Appl Biochem Biotechnol 109:207–225
Franciscon E, Zille A, Fantinatti GF, Silva IS, Cavaco PA, Durrant LR (2009) Microaerophilic–aerobic sequential decolourization/biodegradation of textile azo dyes by a facultative Klebsiella sp. strain VN-31. Process Biochem 44:446–452
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Li C, Liu D, Yang X, Chen S, Deng J, Rong H, Li C (2019) Efficient biodegradation of malachite green by a newly isolated Klebsiella pneumoniae strain WA-1. Environ Prog Sustain Energy. https://doi.org/10.1002/ep13346
Lineweaver H, Burk D (1934) The determination of enzyme dissociation constants. J Am Chem Soc 56:658–666
Misal SA, Lingojwar DP, Shinde RM, Gawai KR (2011) Purification and characterization of azoreductase from alkaliphilic strain Bacillus badius. Process Biochem 46:1264–1269
Moutaouakkil A, Zeroual Y, Zohra Dzayri F, Talbi M, Lee K, Blaghen M (2003) Purification and partial characterization of azoreductase from Enterobacter agglomerans. Arch Biochem Biophys 413:139–146
Ooi T, Shibata T, Matsumoto K, Kinoshita S, Taguchi S (2009) Comparative enzymatic analysis of azoreductases from Bacillus sp. B29. Biosci Biotechnol Biochem 73:1209–1211
Ooi T, Shibata T, Sato R, Ohno H, Kinoshita S, Thuoc TL, Taguchi S (2007) An azoreductase, aerobic NADH-dependent flavoprotein discovered from Bacillus sp.: functional expression and enzymatic characterization. Appl Microbiol Biotechnol 75:377–386
Padamavathy S, Sandhya S, Swaminathan K, Subrahmanyam Y, Kaul S (2003) Comparison of decolorization of reactive azo dyes by microorganisms isolated from various sources. J Environ Sci 15:628–632
Pinheiro HM, Touraud E, Thomas O (2004) Aromatic amines from azo dye reduction: status review with emphasis on direct UV spectrophotometric detection in textile industry wastewaters. Dyes Pigm 61:121–139
Punj S, John GH (2009) Purification and identification of an FMN-dependent NAD(P)H azoreductase from Enterococcus faecalis. Curr Issues Mol Biol 11:59–66
Stolz A (2001) Basic and applied aspects in the microbial degradation of azo dyes. Appl Microbiol Biotechnol 56:69–80
Sudha M, Saranya A, Selvakumar G, Sivakumar N (2014) Microbial degradation of Azo Dyes: a review. Int J Curr Microbiol App Sci 3:670–690
Walker JM (2009) The bicinchoninic acid (BCA) assay for protein quantitation In: The protein protocols handbook. Springer, pp 11–15
Wang CJ, Hagemeier C, Rahman N, Lowe E, Noble M, Coughtrie M, Sim E, Westwood I (2007) Molecular cloning, characterisation and ligand-bound structure of an azoreductase from Pseudomonas aeruginosa. J Mol Biol 373:1213–1228
Wong PK, Yuen PY (1996) Decolorization and biodegradation of methyl red by Klebsiella pneumoniae RS-13. Water Res 30:1736–1744
Zabłocka-Godlewska E, Przystaś W, Grabińska-Sota E (2014) Dye decolourisation using two Klebsiella strains. Water Air Soil Poll 226:2249–2264
Zimmermann T, Kulla HG, Leisinger T (1982) Properties of purified Orange II azoreductase, the enzyme initiating azo dye degradation by Pseudomonas KF46. Eur J Biochem 129:197–203
Acknowledgments
The authors would like to thank Professor Saravanan Matheshwaran, Department of Biological Sciences and Bioengineering, IIT Kanpur, for sharing his laboratory facilities on payment basis. The authors would also like to thank him and his research students for the many meaningful discussions. The authors would also like to thank Professor Sri Sivakumar, Department of Chemical Engineering, IIT Kanpur, for sharing his lab facility ELISA plate spectrophotometer. The authors also thank his student Madhu for the assistance provided while using the spectrophotometer.
Funding
The partial financial support from the Department of Science and Technology, Government of India, New Delhi, India, at the early stages of this work is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Code availability
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Dixit, S., Garg, S. Enzymatic degradation of sulphonated azo dye using purified azoreductase from facultative Klebsiella pneumoniae. Folia Microbiol 66, 79–85 (2021). https://doi.org/10.1007/s12223-020-00824-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12223-020-00824-2