
Abstract
Fifty fluorescent pseudomonads were isolated from rhizospheric soil of green gram from nearby area of Kaziranga, Assam, India and assayed for their extracellular proteinase production. Out of these isolates, 20 were found to be prominent in proteinase production. Genetic diversity of the 20 isolates were analyzed through BOX-PCR fingerprinting and 16S rDNA-RFLP along with three reference strains, viz., Pseudomonas fluorescens (NCIM2099T), Pseudomonas aureofaciens (NCIM2026T), and Pseudomonas aeruginosa (MTCC2582T). BOX-PCR produced two distinct clusters at 56% similarity coefficient and seven distinct BOX profiles. 16S rDNA-RFLP with three tetra-cutters restriction enzymes (HaeIII, AluI, and MspI) revealed two major clusters A and B; cluster A contained only single isolate FPS9 while the rest of 22 isolates belonged to the cluster B. Based on phenotypic characters and 16S rDNA sequence similarity, all the eight highly proteinase-producing strains were affiliated with P. aeruginosa. The proteinase was extracted from two most prominent strains (KFP1 and KFP2), purified by a three-step process involving (NH4)2SO4 precipitation, gel filtration, and ion exchange chromatography. The enzyme had an optimal pH of 8.0 and exhibit highest activity at 60°C and 37°C by KFP1 and KFP2 respectively. The specific activities were recorded as 75,050 (for KFP1) and 81,320 U/mg (for KFP2). The purified enzyme was migrated as a single band on native and SDS-PAGE with a molecular mass of 32 kDa. Zn2+, Cu2+, and Ni2+ ion inhibited the enzyme activity. Enzyme activity was also inhibited by EDTA established as their metallo-proteinase nature.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Microbial extracellular enzymes have attracted the attention from research workers all over the world due to their wide range of physiological, analytical and industrial application. More than 3,000 different microbial extracellular enzymes have been reported so far (Patel et al. 2005). Out of these, proteinases are the most important enzyme from industrial point of view due to their wide variety of functions and various important biotechnological applications (Kamath et al. 2010; Mohen et al. 2005). Microbes serve as the preferred source of proteinase because of their rapid growth, the limited space required for their cultivation, and the ease with which they can be genetically manipulated to generate new enzymes with altered properties (Rao et al. 1998).
Green gram is one of the important pulse crops in India. It is believed that green gram is a native of India and Central Asia and grown in these regions since prehistoric times. Besides an important source of human food and animal feed, green gram also plays an important role in sustaining soil fertility by improving its physical properties and fixing atmospheric nitrogen, on the other hand, pulses are the second most important staple food after rice. In India green gram was grown over on an area of 30,084 ha with a production of 10,532 tons in 2005–2006. So far, no report has been found regarding pseudomonas diversity around green gram rhizosphere in this area.
North–East India is widely known for its rich biodiversity and its untapped bioresearches have been identified as the Indo-Burma Mega hot spot (Myers et al. 2000). The rich microbial diversity of this region has drawn the attention from the researchers. Previously, we reported genetic and functional diversity of antagonistic potential fluorescent pseudomonads isolated from tea rhizosphere of this hot spot (Saikia et al. 2011b). Moreover, we have isolated a potential biocontrol strain Brevibacillus laterosporus from a natural hot water spring of this region (Saikia et al. 2011a). In continuation of the previous work, the objectives of the present work are—(1) isolation and diversity analysis of proteinase producing fluorescent pseudomonads from rhizospheric soil of green gram and (2) purification and characterization of proteinase from fluorescent pseudomonads.
Materials and methods
Soil sampling and isolation of bacteria
Soil samples were collected from different locations of green gram cultivating areas near by Kaziranga National Park. The soil samples were combined and passed through 0.002 m sieve and preserved at 4°C until use. A total of 150 bacteria were isolated from the collected soil samples in King’s B agar (KB) medium and pure cultures were obtained by repeated streaking on KB medium. Pure cultures were screened through UV light (λ = 356 nm) to identify fluorescent pseudomonads. Thus, 50 isolates were selected on the basis of fluorescence and kept in 4°C until use.
Microbial strains
Reference strains of pseudomonads, Pseudomonas fluorescens NCIM2099T and Pseudomonas aureofaciens NCIM2026T were obtained from National Collection of Industrial Microorganisms (NCIM), National Chemical Laboratory, Pune, India and Pseudomonas aeruginosa MTCC2582T was from Institute of Microbial Technology, Chandigarh, India.
Screening for proteinase production
A loopful colony from each of the cultures were spot inoculated on the skimmed milk agar medium (0.5% pancreatic digest casein; 0.25% yeast extract; 0.1% dextrose; 2.8% skimmed milk and 1.5% agar) and incubated at 30 ± 2°C for 24 h. Occurrence of clear proteinolytic zone around the colonies confirmed proteinase producing ability of the screened bacteria (Salwan et al. 2010).
Phenotypic characterization
Bacterial isolates were observed under microscope after gram staining and different morphological as well as biochemical tests such as shape, fluorescent pigments, motility, nitrate reduction, oxidase, methyl red, starch hydrolysis, nitrate reduction, citrate utilization, indole production, and sugar (glucose) utilization test were carried out with five replications (Bergey et al. 1994).
Genotypic analysis
Genomic DNA of 20 fluorescent pseudomonads and three reference strains were extracted by GenElute™ Bacterial Genomic DNA Extraction kit (Sigma, USA). The DNA purity and quantity were checked by spectrophotometer at 260 and 280 nm. The genotypic analyses of 23 Pseudomonas strains were carried out by BOX-AIR1 primer (5′-CTACGGCAAGGCGACGCTGACG-3′) as described earlier (Louws et al. 1994). A 10-μL PCR product together with 500 bp marker (Bangalore Genei, India) was separated using 1.5% agarose gel stained with ethidium bromide in 1 × TAE. A photograph of the gel was taken by gel documentation system (Syngene, Cambridge, UK) and stored as TIFF file for further analysis.
PCR-RFLP analysis of 16S rDNA
Amplification of 16S rDNA region was performed by using bacterial universal primers PA (5′ AGAGTTTGATCCTGGCTAG 3′) and rP2 (5′ ACGGCTACCTTGTTACGACTT 3′) as described earlier (Edwards et al. 1989). Two hundred nanograms of purified PCR products were digested with three units of restriction endonucleases HaeIII, AluI, and MspI (Bangalore Genei, India) as per instructions of the manufacturer. The restriction fragments were analyzed by 2.5% agarose gel in 1× TAE electrophoresis buffer containing 10 μg/ml ethidium bromide at 40 V for 3 h.
Molecular phylogenetic analysis
Eight highly proteinase producing fluorescent pseudomonads strains were identified by 16S rDNA partial sequence. The PCR amplified 16S rDNA were purified by a QIA quick PCR purification kit (Qiagen) and sequenced with the same primers. DNA sequence was determined by fluorescent terminators (Big Dye, Applied Biosystems) and run in an Applied Biosystems ABI prism automated DNA sequencer (3,130 × l). The 16S rRNA gene sequences of bacteria used in the phylogenetic analysis were retrieved from NCBI GenBank.
Effect of carbon and nitrogen sources in enzyme production
Among the isolates, KFP1 and KFP2 showed highest ability of proteinase production, so the rest of the experiments were carried out with these isolates only. The effect of carbon sources in enzyme production was measured replacing glucose by different carbon sources, viz., starch, wheat bran, rice bran, maltose, and sucrose in the enzyme production medium (Kalaiarasi and Sunitha 2009).
To observe the effect of nitrogen sources various nitrogen sources such as beef extract, tryptone, glycine, peptone, and wheat bran were used instead of yeast extract (Kalaiarasi and Sunitha 2009).
Proteinase purification
The bacterial isolates were grown for 48 h at 30°C in proteinase producing medium (g/L—glucose, 10; (NH4)2SO4, 2; K2HPO4, 1; KH2PO4, 1; MgSO4·7H2O, 0.4; MnSO4·H2O, 0.01; FeSO4·7H2O, 0.01; and yeast extract, 1; pH 8.0) with an agitation of 200 rpm. The centrifuged cell free supernatant was precipitated with 70% (NH4)2 SO4. The precipitate was resuspended in 20 mM Tris HCl buffer (pH 8.0) and dialyzed against the same buffer. The precipitate was collected by centrifugation and resuspended in 20 mM Tris-HCl buffer (pH 8.0), and dialyzed against the same buffer. The dialyzed solution was purified through Sephadex G-100 column (2 × 0.7 m) pre-equilibrated with 20 mM Tris-HCl. Fractions (5 ml) were collected individually and those showing proteinase activity were resolved on a DEAE-cellulose column (2 × 0.095 m) equilibrated with 20 mM Tris-HCl (pH 8.0). The enzymes were eluted with a linear gradient of 0–0.75 mol/L NaCl in the same buffer. Active fractions were concentrated by lyophilization for further work. All steps were carried out at 4°C.
Proteinase assay
Proteinase activity was determined according to Shimogaki et al. (1991). Enzyme units were measured by using tyrosine (0–100 μg) as standard. One unit of proteinase activity was defined as the amount of enzyme required to produce 1 μg of tyrosine/min. Protein concentrations of purified active fractions were determined according to Lowry with bovine serum albumin as a substrate. Enzyme was subjected to electrophoresis in a 15% SDS-PAGE gel and stained with Coomassie Brilliant Blue to determine the purity and molecular mass of the enzyme. Activity staining of purified proteinase was performed by gelatin native PAGE zymography (Huessen and Dowdle 1980) using 10 mg/ml gelatin in 10% polyacrylamide mixture.
Effect of temperature and pH on alkaline proteinase activity
The influence of temperature on the catalytic activity of purified alkaline proteinase was determined by measuring the activity at temperature range from 20°C to 90°C under standard assay condition (Vazquez et al. 2004). Effect of pH was determined by using pH solution with range from 5.0 to 12 (Vazquez et al. 2004) in the presence of different substrates, i.e., casein, azocasein, and gelatin.
Effect of inhibitors and metal ions
EDTA, PMSF, 1,10-phenanthroline (10 mM each) and different metal ions, viz., Zn2+, Cu2+, and Ni2+ (1 mM each) were tested for ability to inhibit proteinase activity. Enzymes were incubated with each reagent at 37°C for 20 min (pH 8.0). The residual activity was measured using casein as a substrate (Vazquez et al. 2004).
Data analysis
For the analysis of 16S rDNA-RFLP data, the character state “1” was given for a band, which could be clearly detected in the gel and “0” was assigned if it was absent or it was not possible to determine. The data matrix thus generated was calculated by Jaccard’s similarity coefficient. Each pair-wise comparison was constructed from the similarity matrix by the unweighted pair group method with arithmetic mean (UPGMA). For BOX-PCR fingerprinting analysis, the photograph (Fig. 2) was imported into the software package BioNumerics version 2.5 (Applied Maths, Belgium). Similarity matrices of densitometric curves of the gel tracks were calculated using the Pearson’s product moment correlation coefficient followed by tree construction using UPGMA algorithm. Phylogenetic and molecular evolutionary analyses with the sequences of eight strains were conducted by using software included in MEGA4. All the eight sequences of 16S rDNA were aligned using the multiple sequence alignment program CLUSTAL-W. The pair wise evolutionary distances matrix was generated (Jukes and Cantor 1969) and phylogenetic tree was inferred by the Maximum-parsimonious phylogenetic consensus tree method. Tree topologies were evaluated by boot strap analysis (Felsenstein 1985) based on resamplings of 1,000 times of the neighbor joining data set. The sequences were submitted to the NCBI Gene Bank. Data generated from biochemical characterization of proteinase were analyzed by means of one-way ANOVA and means were compared by the Tukey’s test, using the SPSS software (ver. 10.1, SPSS Inc., www.spss.com).
Results
Morphological and biochemical characterization
Out of 50 fluorescent pseudomonads, 20 isolates showed positive in proteinase production. Of the 20 isolates, eight highly proteinase producing fluorescent pseudomonads were further characterized based on morphological and biochemical characters, recorded as rod shaped, motile and showed positive in nitrate reduction, oxidase test, citrate utilization, and sugar (glucose) utilization. The isolates were found to be negative in gram reaction, methyl red test, starch hydrolysis and indole production (see Electronic supplementary materials (ESM) 1)
Genotypic analysis
The cluster analysis with respect to the complex amplified banding pattern of BOX-PCR resulted two distinct genomic clusters at a 56% similarity coefficient and generated seven BOX profiles (Fig. 1). Profile I comprised three isolates viz., FPS26, NAM7, and KFP4; profile II consisted of highest numbers (eight numbers) of isolates, i.e., FPS14, FPS18, FPS5, FPS37, FPS24, KFP8, KFP3, and a reference strain P. aeruginosa MTCC2582T; profile III comprised three isolates, KFP5, KFP9, and FPS9; and profile IV contained four isolates, KFP7, FPS211, FPS357, and FPS22. Similarly, profile V included three isolates viz., FPS324, KFP1, and KFP2. However, profiles VI and VII consisted of two reference strains (P. fluorescens NCIM 2099T and P. aeurofaciens NCIM 2026T) only.

Cluster analysis of BOX-PCR profiles showing the genotypic diversity of proteinase producing fluorescent pseudomonads with comparison to the three reference strains (P. fluorescens NCIM 2099T, P. aeruginosa MTCC 2582T, and P. aureofaciens NCIM 2026T). Dendrogram was obtained from the similarity coefficient (Jaccard Coefficient) and clustering was done by UPGMA
PCR-RFLP analysis
The 16S rDNA RFLP analysis with three tetra-cutters restricted enzymes (HaeIII, AluI, and MspI) revealed significant genetic diversity among the 20 isolates (see ESM 2). HaeIII showed ten restriction patterns; whereas AluI and MspI revealed 8 and 13 restriction patterns, respectively. Except for isolate FPS9 and the reference strains P. fluorescens NCIM 2099T and P. aureofaciens NCIM 2026T, all other isolates showed a common banding pattern at 580 bp after digestion with HaeIII. Similarly, banding pattern at 200 bp was common to all other isolates except FPS9. Restriction fragments at 120 and 180 bp were also identical to all the isolates including the reference strains. FPS9 had unique banding at 300 and 320 bp.
Digestion with AluI produced similar banding pattern for FPS9 and P. fluorescens NCIM 2099T at 600 bp, whereas a single restriction pattern was observed for FPS9 at 580 bp. Except for isolate FPS9, the banding pattern at 400 bp was common to all other isolates and P. fluorescens NCIM 2099T. Similarly, common band was also observed at 200 and 190 bp for all other isolates except FPS9.
Among the restriction enzymes used in this study, MspI gave highest (13 numbers) restriction patterns. Isolates FPS324 and KFP7 revealed common fragments at 1,000 bp; similarly, fragments at 900 bp were common for KFP5, FPS324, and KFP7. Only isolate KFP7 showed banding at 700 and 600 bp. Common restriction fragments was also recorded at 580 bp for all other isolates except FPS26, FPS9, and FPS324.
A critical analysis of the dendrogram obtained from the distance matrix UPGMA revealed two main clusters A and B at 37% similarity coefficient value (Fig. 2). Cluster A contained the single isolate FPS9, whereas rest 22 isolates (including reference strains) were under cluster B. Cluster B further sub-divided into two sub-clusters C and D at 77% similarity coefficient value. Sub-cluster C comprised of single isolate, KFP7. At 81% similarity coefficient, sub-cluster D was further divided into sub-clusters E and F; E contained two reference strains, P. aeurofaciens NCIM 2026 and P. fluorescens NCIM 2099. The sub-cluster F was again subdivided into two sub-clusters G and H at 83% similarity coefficient. Sub-cluster G consisted of two isolates, FPS324 and KFP5 with 86% similarity. Seventeen isolates along with reference strain P. aeruginosa MTCC 2582T were belonged to the sub-cluster H.

Combined dendrogram of proteinase producing fluorescent pseudomonads isolates with comparison to the reference strains (P. fluorescens NCIM2099T, P. aeruginosa MTCC2582T, and P. aureofaciens NCIM 2026T based on cluster analysis of 16S rDNA-RFLP products with restriction endonuclease HaeIII, AluI, and MspI using the UPGMA algorithm and the Jaccard coefficient
Phylogenetic analysis
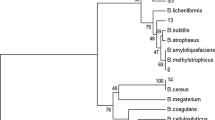
Blast analysis of the 16S rDNA sequence (NCBI accession no. HQ007938 to HQ007945) of eight isolates—KFP1, KFP2, KFP3, KFP4, KFP5, KFP7, KFP8, and KFP9 were affiliated with P. aeruginosa (see ESM 1). A comparative analysis of 16S rRNA gene of the P. aeruginosa strains was performed by constructing maximum-parsimonious phylogenetic consensus tree with reference sequences from the NCBI GenBank database presented in the Fig. 3. The consensus tree inferred two parasimonious trees, three monophyletic group and three single taxa at 75% consensus tree cut-off value. Strains KFP5 and KFP7 are similar to each other supporting 100% bootstrap value. Strain KFP3 formed a clade with P. aeruginosa (HQ202540), with 100% recovery in bootstrap analysis. However, strain KFP8 constitutes a single taxa opposing relationship with any other group. The Closest similarity of sequence of four P. aeruginosa, i.e., KFP1, KFP2, KFP4, and KFP9 isolates were found to be uncultured Pseudomonas sp. (GQ417590), uncultured bacterium clone P6D102-474(EF510076), uncultured bacterium clone 16sps24-2b04.plk(FM997202), uncultured bacterium clone P6D102-457(EF510087), respectively (see ESM 1). The closest neighbours of some of the strains belong to uncultured bacterium clones representing their lack of taxonomic standing.

Maximum parsimonious phylogenetic consensus tree derived from 16S rDNA partial sequence data, showing the affiliation of P. aeruginosa. The 16S rDNA sequence of Escherichia coli (J01859) was chosen arbitrarily as the outgroup sequence for the trees. Numbers at internal nodes are bootstrap support values (in percent)
Effect of carbon and nitrogen sources on proteinase production
In this study, various carbon sources (1%) such as starch, maltose, sucrose, rice bran and wheat bran were used to replace the original carbon source (glucose) in the growth medium. Results obtained (Fig. 4a) showed that both the strains were able to utilize most of the carbon sources tested with highest enzyme production in wheat brand. Similarly, in case of nitrogen sources, among beef extract, tryptone, glycine, peptone, and wheat brand; maximum enzyme production also recorded in presence of wheat bran (Fig. 4b). However enzyme production in KFP1 was higher than KFP2 in all the carbon and nitrogen sources used. Enzyme production by KFP1 was significantly higher than KFP2 while taken sucrose as carbon source.

Effect of carbon and nitrogen sources on proteinase activity; a effect of carbon sources and b effect of nitrogen sources
Partial purification of proteinase
Protein precipitation by 70% ammonium sulphate yielded 83% and 82% of recovery for the strains KFP1 and KFP2, respectively (Table 1). The precipitate obtained after centrifugation at 10,000 rpm at 4°C was dissolved in 20 mM Tris-HCl (pH 8.0). Purification by sephadex column results 53% (KFP1) and 52% (KFP2) recovery. The final step of purification with ion exchange chromatography (DEAE—cellulose column) in the presence of 0.16 mol/L NaCl revealed a single peak of proteinase activity. The purified enzyme was found to be homogenous by 15% SDS-PAGE and the molecular mass of proteinase as estimated from SDS-PAGE was about 32 kDa for both the enzyme samples (Fig. 5a). Zymogram activity staining also revealed clear single band of proteinolytic activity (Fig. 5b).

a SDS-PAGE (15%) of the purified proteinase from KFP1 and KFP2; M, molecular mass standard; L1, purified proteinase of KFP1; and L2, purified proteinase from KFP2 showing 32 kDa molar mass. b Zymography of purified proteinase; L1, zymography of KFP1 purified proteinase and L2, zymography of KFP2-purified proteinase
Biochemical characterization of the proteinase
The optimum temperature for the enzyme activity was recorded at pH 8.0. Enzyme of the strain KFP1 showed optimum temperature at 60°C and KFP2 at 37°C. Enzymes from both the strains were found to be almost stable at 80°C as 70% and 66% activity was recorded at this temperature for the strains KFP1 and KFP2, respectively. Beyond 80°C the enzyme activity was abruptly decreased (Fig. 6a). Effect of different pH was measured at temperature 37°C. Maximum enzyme activity for KFP1 was recorded at pH 8.0 for each substrate used (Fig. 6b). Proteinase of KFP2 also showed maximum activity at pH 8.0 while casein and gelatin were used as substrate; however in case of azocasein as a substrate, optimum activity was recorded at pH 6.0 (Fig. 6c). Activity was stable i.e., 90% of the activity was recorded at pH 9.0 for the proteinase and beyond pH 9.0 the enzyme activity reduced rapidly while used casein and gelatin as substrate. In case of KFP2 enzyme activity falls rapidly after pH 6.0 using azocasein as a substrate (Fig 6c). From statistical analysis of the data from both experiments revealed that at P < 0.05 level, mean values for relative enzyme activity were not significantly different with response to the range of temperature from 30°C to 80°C but significantly different in various pH ranges.

Effect of temperature and pH on proteinase activity; a effect of temperature, b effect of pH on proteinase activity of KFP1, and c effect of pH on proteinase activity of KFP2
The effects of chemical inhibitors and metal ions are summarized in Table 2. Both EDTA and 1,10-phenanthroline inhibited the proteinase activity of KFP1. The enzyme activity of KFP2 was inhibited by EDTA, but insensitive to 1,10-phenanthroline. However, enzyme activity was insensitive to PMSF. The enzyme activity was found to be inhibited by metal ion, Zn2+, Cu2+, and Ni2+ (Table 2).
Discussion
Phylogenetic analysis through 16S rDNA sequence analysis, suggested dominance of P. aeruginosa in green gram rhizosphere among proteinase producing fluorescent pseudomonads. A comparative diversity study of all the proteinase producing strains through 16S rDNA-RFLP by three tetra cutter endonucleases HaeIII, AluI, and MspI and BOX-AIR primer analysis with respect to the reference strains revealed significant difference among them. The BOX-PCR genomic fingerprints confirmed intra specific diversity among the strains. The random distribution and occurrence of fluorescent pseudomonads strains from green gram rhizosphere of different locations into various 16S rDNA-RFLP and BOX-PCR profile indicate the lack of locations specific diversity in the rhizospheric soil of green gram. The genetic variation among the proteinase producing prominent strains might be due to mutation (Oliver et al. 2000) and other genetic variations like recombination (Ochman et al. 2000). Habitat variability for a variety of soil properties also may be positively correlated with genetic diversity in natural populations of a soil bacterium (Mc Arthur et al. 1988). Differences in frequency and intensity of competitive interactions among the locations of isolation could also influence genetic composition and diversity of bacteria (Davelos et al. 2004).
In this study, the activities of both proteinase was found to be almost stable at 70°C temperature which reflects their thermo resistively nature, however showed different temperature optima. Variation in optimum pH by using the different substrates for proteinase of KFP2 explained the fact that these substrates are proteins exhibiting the states of protonation and deprotonation, which may alter the bonds between the substrate and the catalytic site and consequently affect enzyme catalysis. Variation of enzyme production yield was not constant for both organisms at selected range of temperature and pH. Similarly, significant difference in enzyme production was noticed with different carbon as well as nitrogen sources. These differences may be attributed to influence of different metabolism aspects of the two bacterial species. The maximum growth of the two strains found in wheat bran as both carbon and nitrogen sources. Since cheaper sources of raw materials is the one of the most prevalent factor for commercialization of the production processes, hence ability of the microbial agent to grow and produce enzymes using these sources may be promising (Kanekar et al. 2002).
SDS-PAGE analysis revealed the molar mass of the enzymes as 32 kDa. Activities of both the enzymes were inhibited by EDTA, indicating that they are metallo-proteinase. The enzyme activity of KFP1 was found to be inhibited by 1,10-phenanthroline, however, the enzyme activity of KFP2 was not sensitive to 1,10-phenanthroline, differentiating them from one another. Though proteinase activity of KFP1 was totally inhibited by EDTA, however, KFP2 retaining 32% of residual activity in presence of EDTA. Similarly there was difference in inhibition of enzyme activity in the presence of Cu2+ metal ion, while comparing proteinase from both the strains. The differences in inhibitor’s mode of action may be correlated to the degree of reduction of some residues near the active motif.
Overall study demonstrated that—(1) a significant genetic diversity was observed among the proteinase producing fluorescent pseudomonads strains in green gram rhizosphere and (2) being thermo-stable and high pH tolerant, the two metallo-proteinase can be considered for the detergent industry.
Abbreviations
- EDTA:
-
Ethylenediaminetetraacetic acid
- PMSF:
-
Phenylmethylsulfonylfluoride
- TAE:
-
Tris-acetate ethylenediaminetetraacetic acid
- DEAE:
-
Diethylaminoethyl
- SDS-PAGE:
-
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
References
Bergey DH, Holt JG et al (1994) Bergey's manual of determinative bacteriology. Williams & Wilkins, Baltimore
Davelos AL, Xiao K, Samac DA, Martin AP, Kinkel LL (2004) Spatial variation in Streptomyces genetic composition and diversity in a Prairie soil. Microb Ecol 448:601–612
Edwards U, Rogall TH, Blocker H, Emde M, Bottger EC (1989) Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding for 16S ribosomal RNA. Nucleic Acids Res 17:7843–7853
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Huessen C, Dowdle EB (1980) Electrophoretic analysis of plasminogen activators in polyacrylamide gels containing sodium dodecyl sulfate and copolymerized substrates. Anal Biochem 102:196–202
Jukes TH, Cantor CR (1969) Evolution of protein molecules. In: Munro HN (ed) Mammalian protein metabolism, vol 3. Academic, New York
Kalaiarasi K, Sunitha P (2009) Optimization of alkaline protease production from Pseudomonas fluorescens isolated from meat waste contaminated soil. Afr J Biotechnol 8:7035–7041
Kamath P, Subrahmanyam VM, Venkata Rao J, Vasanth Raj P (2010) Optimization of cultural conditions for protease production by a fungal species. Indian J Pharm Sci 72:161–166
Kanekar PP, Nilegaonkar SS, Sarnaik SS, Kelkar AS (2002) Optimization of protease activity of alkaliphilic bacteria isolated from an alkaline lake in India. Biores Technol 85:87–93
Louws FJ, Fulbright DW, Stephens CT, de Bruijn FJ (1994) Specific genomic fingerprints of phytopathogenic Xanthomonas and Pseudomonas pathovars and strains generated with repetitive sequences and PCR. Appl Environ Microbiol 60:2286–2295
Mc Arthur JV, Kovacic DA, Smith MH (1988) Genetic diversity in natural populations of a soil bacterium across a landscape gradient. Proc Natl Acad Sci USA 85:9621–9624
Mohen FN, Dileep D, Deepthi D (2005) Potential application of protease isolated from Pseudomonas auriginosa PD100. Biotechnol Ind 8:197–203
Myers N, Russel AM, Cristina G, Gustavo-Foneca AB, Kent J (2000) Biodiversity hotspots for conservation priorities. Nature 403:853–858
Ochman H, Lawrence JG, Groisman EA (2000) Lateral gene transfer and the nature of bacterial innovation. Nature 405:299–304
Oliver A, Canton R, Campo P, Baquero F, Blazquez J (2000) High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288:1251–1254
Patel R, Dodia M, Singh S (2005) Extracellular alkaline protease from a newly isolated haloalkaliphilic Bacillus sp.: production and optimization. Process Biochem 40:3569–3575
Rao MB, Tanksale AM, Ghatge MS, Deshpande VV (1998) Molecular and biotechnological aspects of microbial protease. Microbiol Mol Biol Rev 62:597–635
Saikia R, Gogoi DK, Mazumder S, Yadav A, Sarma RK, Bora TC, Gogoi BK (2011a) Brevibacillus laterosporus strain BPM3, a potential biocontrol agent isolated from a natural hot water spring of Assam, India. Microbiol Res 166:216–225
Saikia R, Sarma RK, Yadav A, Bora TC (2011b) Genetic and functional diversity among the antagonistic potential fluorescent pseudomonads isolated from tea rhizosphere. Curr Microbiol 62:434–444
Salwan R, Gulati A, Kasana R (2010) Phylogenetic diversity of alkaline protease-producing psychrotrophic bacteria from glacier and cold environment of Lahaul and Spiti, India. J Basic Microbiol 50:150–159
Shimogaki H, Takeuchi K, Nishino T, Ohdera M, Kudo T, Ohba K, Iwama M, Irie M (1991) Purification and properties of a novel surface active agent and alkaline-resistant protease from Bacillus sp. Agric Biol Chem 55:2251–2258
Vazquez S, Coria S, Cormack W (2004) Extracellular protease from eight psychrotolerant Antarctic strains. Microbiol Res 159:157–166
Acknowledgments
The work is supported by a Network Project (AMAAS), sponsored by Indian Council of Agricultural Research (ICAR), Government of India, New Delhi. The authors are thankful to the funding agency. The authors are also thankful to P.G. Rao, Director, North-East Institute of Science and Technology (CSIR), Jorhat, India for providing necessary facilities to carry out the work.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary materials
Below is the link to the electronic supplementary material.
Supplementary material 1
Phenotypic and molecular traits of fluorescent pseudomonads (Pseudomonas aeruginosa) (DOC 39 kb)
Supplementary material 2
Fig. a, b, c Restriction patterns of PCR amplified fragment of 16S rDNA digested with HaeIII, AluI, and MspI, respectively. Lane name with respective isolates: M, 100 bp molecular marker; 1, FPS 26; 2, KFP 5; 3, FPS 9; 4, FPS 324; 5, FPS 14; 6, KFP 7; 7, KFP8; 8, FPS 37; 9, NAM 7; 10, FPS 22; 11, FPS 211; 12, KFP1; 13, FPS 5; 14, FPS 24; 15, KFP3; 16, KFP 9; 17 KFP 2; 18, KFP 357; 19, FPS 18; 20, KFP4; 21, P. fluorescens NCIM2099T; 22, P. aeruginosa MTCC2582T; and 23, P. aureofaciens NCIM 2026T (DOC 180 kb)
Rights and permissions
About this article
Cite this article
Sarma, R.K., Debnath, R., Saikia, R. et al. Phylogenetic analysis of alkaline proteinase producing fluorescent pseudomonads associated with green gram (Vigna radiata L.) rhizosphere. Folia Microbiol 57, 129–137 (2012). https://doi.org/10.1007/s12223-012-0097-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12223-012-0097-6