
Abstract
After a short introduction to the general phenomenon of chirality, the implications for synthesis and application of chiral drugs are discussed. In a first part, the historical development is briefly described. Up to the 1950s, most medications were either of natural origin, or made semi-synthetically from natural products. In these cases, only one enantiomer was usually present, i.e., the drugs were used as single enantiomers. This changed when totally synthetic drugs began to dominate the market, since these drugs were usually prepared, tested and applied as racemates. Due to the observation of negative effects such as the Thalidomide (Contergan) tragedy, stricter regulations were introduced and as a consequence, chiral drugs are now almost exclusively applied as single enantiomers. In a second part, the challenges for the industrial synthesis of chiral drugs are discussed. The various approaches to prepare enantiomerically pure compounds are briefly described and discussed in the context of industrial process development. Important for the choice of the production technology are criteria such as time constraint, complexity of the molecule, cost considerations and maturity of the technology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Background
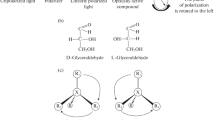
Chirality is an all-encompassing phenomenon (Blaser et al. 2012). In nature, both macroscopic as well as microscopic objects can be chiral. In molecular terms, chirality is a geometric property of a particular sub-class of stereoisomers. A molecule is termed chiral, when it can exist in two forms (the enantiomers) which have the same chemical structure but are mirror images of each other which are non-superimposable. The concept of chirality has played an essential role in the development of stereochemical models, but more importantly, it has a very strong influence on our daily life in the context of the application of chiral bioactive compounds such as pharmaceuticals, agrochemicals or flavors and fragrances.
Although nature frequently exhibits a high degree of symmetry in terms of general morphology, at the molecular level the natural world is highly asymmetric. Enzymes, proteins, polysaccharides, nucleic acids, and many other basic components of plants and animals are chiral and occur in enantiopure form. The implications of the chiral nature on the properties of biological systems are profound. For example, enzymes or proteins will distinguish between the two enantiomers of a chiral drug leading to sometimes dramatically different effects. This can be understood by imagining an enzyme as having a glove-like cavity that binds a substrate. If the glove is right-handed, then one enantiomer will fit inside and be bound, whereas the other enantiomer will have a poor fit and is unlikely to bind. Similarly, since our sensory receptors involved in taste and smell are also chiral, enantiomers of chiral compounds often taste and smell differently. For example, natural l-asparagine is bitter, whereas artificial d-asparagine is sweet.
Even though most of these facts have been known for a long time, according to Gal (2006) one can distinguish different phases in the history of chiral drugs. For thousands of years and until the beginning of the nineteenth century most remedies were used as crude plant extracts without any clue as to the nature or identity of the active ingredient(s) within, let alone any understanding of the chirality of the molecules involved. A milestone was the realization by Biot of a molecular-structural cause of optical rotation; coupled with his discovery in 1815 of the optical rotation of (+)-camphor (Fig. 1), a therapeutic agent, may be considered the earliest scientific hint for chirality in drugs.

Structures of (+)-camphor, morphine and heroin
As long as most remedies were either natural products or semi-synthetic variations thereof, all chiral drugs were in essence single enantiomers. An early example is heroin, a potent opiate narcotic produced from morphine by diacetylation, which was introduced into medical practice in 1898 as a cough suppressant. Heroin may have been the first synthetic single enantiomer drug introduced in clinical medicine. The drug was touted as a “non-addicting” morphine analog in the (obviously mistaken) opinion that it could safely replace morphine and thereby eliminate the latter’s addiction problem.
The situation changed significantly when, starting in the 1950s, fully synthetic drugs began to represent a major segment of new therapeutic agents. For example, the vast majority of synthetic chiral drugs introduced in 1987, ca. 88 %, were racemic. According to Gal, this lack of interest in chirality from the industry may have been the result of a lack of interest in chirality from governmental drug-regulatory agencies. Until 1987, the FDA did not explicitly require the inclusion of information on the enantiomer composition of chiral substances in new drug applications.
Clearly, if a chiral molecule is directed towards a biological target, the two enantiomers should be viewed as distinct compounds that are capable of acting in different ways. The potency, absorption, transport, degradation, and excretion of the two enantiomers can be quite different within the body. Although this may often not be a problem, in the worst scenario the unwanted enantiomer can be highly toxic. A particularly tragic case in point was the sedative Thalidomide (also named Contergan, Fig. 2), which was sold as a racemate and in the late 1950s led to severe malformations in children. It was later found that this was due to the teratogenic nature of the (S)-enantiomer. This case and other observations led to an increased pressure for enantiomerically pure compounds even though it was found that for Thalidomide, applying only the (R)-enantiomer would not have helped, since racemization readily occurs in the body (Muller 1997).

Structure of Thalidomide (Contergan)
Since the 1990s there is a clear trend to develop single enantiomer drugs. In 1988, the FDA announced a set of guidelines addressing these stereochemical topics in relation to the submission of new drug applications (de Camp 1989). As a consequence, the use of racemates has drastically been reduced (see Fig. 3) and an analysis carried out in 2006 by scientists from AstraZeneca, GlaxoSmithKline and Pfizer confirmed this trend: of the 128 compounds then under development in the three companies, 69 (54 %) were molecules containing at least one stereogenic centre, and of the 69 chiral molecules, 67 were being developed as single enantiomers, and only two as racemates (Carey et al. 2006). For several years, the “chiral switch” as (limited) strategy to improve profile (and prolong patent life) of established racemic drugs was quite popular for both originators as well as competitors (Hutt and Valentova 2003). A racemic or chiral switch may be defined as the development of a single enantiomer from a previously marketed racemate. However, not all these re-evaluations have resulted in the expected therapeutic benefits and unpredicted adverse reactions have resulted.

Proportion of chiral drugs approved as racemate
2 Synthesis of single enantiomers in an industrial context
Up to the 1960s, the efficient synthesis of enantiopure chiral chemicals was a very difficult endeavor since few enantioselective synthetic methods were known. Therefore, most synthetic drugs were synthesized as racemates and either tested and used as such or in rare cases resolved via crystallization. In the 1970s, the development of enantioselective methodologies started in earnest and now there is a plethora of enantioselective methods available. Four general approaches for producing enantiopure (ee > 99 %) or enantioenriched compounds have evolved (for a more in-depths description see Blaser et al. 2012).
-
1.
Resolution of racemates via separation of the two enantiomers. This can be achieved via classical crystallization of diastereomeric adducts (usually salts), HPLC on a chiral stationary phase using moving simulated bed technology or by (catalytic) kinetic resolution.
-
2.
The chiral pool approach using chiral building blocks originating from natural products for the construction of the final molecule.
-
3.
Stoichiometric enantioselective syntheses, performed either with chiral reagents or with the help of covalently bound chiral auxiliaries (often from the chiral pool) which render the reactions diastereoselective.
-
4.
Enantioselective catalysis where achiral starting materials are transformed to enantioenriched products with the help of chiral catalysts. Effective catalysts are either synthetic (often called chemocatalysis) or of natural origin (often termed biocatalysis).
It has to be pointed out that the development of an effective new synthesis for even a simple chiral molecule can be quite time consuming and tedious (and without guarantee of success). When developing a process for a new chemical entity (NCE) in the pharmaceutical industry, time restraints can be severe (see Fig. 4) (Blaser 2012). In many cases, it is more important to find a competitive process on time than an optimal process too late. So-called second generation processes, e.g., for chiral switches, for generic pharmaceuticals or the manufacture of other fine chemicals have different requirements; here the time factor is usually not so important but a high performance process is necessary.

Schematic presentation of the development process for a new chemical entity in the pharmaceutical industry
The choice of the optimal method to prepare a chiral target molecule will depend on a number of considerations such as the nature of the chemical reaction, the goal of the synthesis, the know-how of the investigators, the time frame, the available manpower and equipment, and so on. No matter which option will be chosen, the following criteria will influence the selection:
-
Maturity of the enantioselective step. What level of enantioselection can be expected, how well are scope and limitations known?
-
Commercial availability of both enantiomers of the chiral auxiliary. For industrial applications this means that, e.g., chiral ligands must be available on kg scale.
-
Cost (for industrial applications) and effort needed for the over-all synthesis.
-
Familiarity and experience of the investigators with a particular methodology or transformation.
-
Last but not least, the time available for developing the final process.
As a general rule, synthesis of racemic compounds followed by chromatographic resolution is usually the method of choice when a chiral enantiopure product has to be synthesized fast and in small amounts such as in the discovery phase of pharmaceuticals. As the compilation by Carey et al. (2006) indicates, the preferred method for the large scale synthesis of enantiopure drugs was the use of commercially available building blocks (used in 55 % of the 69 discussed syntheses). Resolution of racemic compounds was applied in 28 %, and asymmetric synthesis was used in only 10 % of all cases. One has to realize that process development will be much more complex and time consuming for making single enantiomer drugs, especially when catalytic enantioselective processes are envisaged. Nevertheless, there are a growing number of cases where enantioselective processes are actually used for the production of chiral drugs (Blaser et al. 2008) and this number should (hopefully) increase in the future (Busacca et al. 2011).
3 Conclusions
It is now established that the two enantiomers of a chiral drug have to be treated as two different bioactive ingredients. Only when the biological profile of the enantiomers is the same will the application of the racemate be approved by the regulatory authorities. The majority of the single enantiomer drugs are produced either by the use of commercial chiral building blocks or via the resolution of racemates. Enantioselective and especially catalytic methodologies are at the moment less often applied but especially for generics there is a growing tendency to apply such modern technologies.
References
Blaser HU (2012) Industrial asymmetric hydrogenation. In: Crawley ML, Trost BM (eds) Applications of transition metal catalysis in drug discovery and development. Wiley, New York, pp 315–342
Blaser HU, Pugin B, Spindler F (2008) Asymmetric catalysis—industrial processes. In: Horvath IT (ed) Encyclopedia of Catalysis (on-line), Wiley-Interscience, 2nd edn, Accessed 20 Nov 2012. http://onlinelibrary.wiley.com/doi/10.1002/0471227617.eoc025/abstract
Blaser HU, Pfaltz A, Wennemers H (2012) Chiral compounds. In: Ullmann’s Encyclopedia of Industrial Chemistry. On-line edition. Accessed 20 Nov 2012. http://onlinelibrary.wiley.com/doi/10.1002/14356007.a18_177.pub2/abstract
Busacca CA, Fandrick DR, Song JJ, Senanayakea CH (2011) The growing impact of catalysis in the pharmaceutical industry. Adv Synth Catal 353:1825–1864
Carey JS, Laffan D, Thomson C, Williams MT (2006) Analysis of the reactions used for the preparation of drug candidate molecules. Org Biomol Chem 4:2337–2347
de Camp WH (1989) The FDA perspective on the development of stereoisomers. Chirality 1:2–6
Gal J (2006) Chiral drugs from a historical point of view. In: Francotte E, Lindner W (eds) Chirality in drug research. Wiley-VCH, Weinheim, pp 3–26
Hutt AT, Valentova J (2003) The chiral switch: the development if single enantiomer drugs from racemates. Acta Facultatis Pharmaceuticae Universitatis Comenianae, Tomus L, pp 7–23
Muller GW (1997) Thalidomide: from tragedy to new drug discovery. Chemtech 27(1):21–25
Author information
Authors and Affiliations
Corresponding author
Additional information
This contribution is the written, peer-reviewed version of a paper presented at the conference “Molecules at the Mirror—Chirality in Chemistry and Biophysics”, held at Accademia Nazionale dei Lincei in Rome on October 29–30 2012.
Rights and permissions
About this article
Cite this article
Blaser, HU. Chirality and its implications for the pharmaceutical industry. Rend. Fis. Acc. Lincei 24, 213–216 (2013). https://doi.org/10.1007/s12210-012-0220-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12210-012-0220-2