
Abstract
To understand the potential association of heat stress resistance with HspB1 induction by aspirin (ASA) in chicken myocardial cells, variations of HspB1 expression and heat stressed-induced damage of myocardial cells after ASA administration were studied in primary cultured myocardial cells. Cytopathological lesions as well as damage-related enzymes, such as creatine kinase-MB (CK-MB) and lactate dehydrogenase (LDH), indicated the considerable protective ability of ASA pre-treatment against acute heat stress. Immunostaining assays showed that heat stress caused HspB1 to relocate into the nucleus, while ASA did not. ELISA analysis, revealed that HspB1 expression induced by ASA averaged 45.62-fold higher than that of the control. These results indicated that the acute heat-stressed injuries were accompanied by comparatively lower HspB1 expression caused by heat stress in vitro. ASA pre-treatment induced a level of HspB1 presumed to be sufficient to protect myocardial cells from acute heat stress in the extracorporal model, although more detailed mechanisms will require further investigation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The animal industry has suffered great economic losses due to the seasonal climate in many regions (Gao et al. 2013; Rhoads et al. 2010). Current studies have indicated that economic losses across several stock farming industries will be quite considerable without primary protection from heat. Annual losses in the USA alone was estimated at $2.4 billion due to non-protection from heat exposure ($897 million, $369 million, $299 million, and $128 million for dairy, beef, swine, and poultry industries, respectively) (N. R. St-Pierre* 2003). Heat stress leads to dysfunction in multiple organs, including the heart, one of the most important organs in multicellular organisms (Yan et al. 2009). Such myocardial injury is believed to be responsible for sudden death induced by heat stress (Ellis 1972; Scheers-Masters et al. 2004). Even in animals that survive the heat stress, acute intracorporal damage would affect the breeding industries. In fact, due to feather cover, lack of perspiratory glands and fast growth in modern commercial breeding facilities, poultry species, such as broiler chickens are more defenseless to high temperatures than mammals (N. R. St-Pierre* 2003; Piestun et al. 2013). Many important organs displayed damages in chickens after acute heat stress exposure, including the heart (Yan et al. 2009). Such injuries will change the permeability of the myocardial cell membrane and cause the release of a series of enzymes such as creatine kinase-MB (CK-MB) and lactate dehydrogenase (LDH) (Saravanan et al. 2013). Increases of these myocardial cell damage-related enzymes have been clearly detected after heat stress (Tang et al. 2013; Yan et al. 2009). Moreover, heat stress exposure leads to obvious pathological changes. The myocardial cells of chickens in our recent studies showed such changes as swollen in size, acute granular or vacuolar degeneration, and karyopyknosis (Wu et al. 2015). All these injuries are indicative of the harmful effects that heat stress would cause to the breeding industries. Thus, developing a feasible solution to protect animals, especially poultry species such as chickens, from heat stress is an important aim.
A natural stress resistance regulatory pathway known as the heat shock response (HSR) is present in many different species. This regulation is mediated by a group of heat shock proteins (Hsps). As studies of Hsps develop, greater attention has been placed on a special class of small heat shock proteins (sHsps) due to their specific protective functions. About ten sHsps (HspB1-HspB10) have been discovered thus far. HspB1, also known as Hsp27, was confirmed to participate in different types of stress resistance without organ specificity. HspB1 was also reported to confer additional protection by associating with the cytoskeleton as large oligomers or by phosphorylation as monomers, thus providing greater cellular protection than any other Hsps (Benn et al. 2002; de Graauw et al. 2005; Kato et al. 1994; Lambert et al. 1999). The protective function of HspB1 in myocardial cells was reported previously by Liu et al. In that study, overexpression of HspB1 was shown to attenuate cardiac dysfunction in transgenic mice (Liu et al. 2007). Based on those findings, it is reasonable to consider that myocardial cell injury possibly can be suppressed if sHsps, especially HspB1, can be induced or overexpressed appropriately.
Aspirin (i.e., acetyl salicylic acid or ASA) was reported to be able to induce different Hsps in various species (Amberger et al. 1999; Sandoval-Montiel et al. 2013), including HspB1 (Ebert et al. 2005). Other studies illustrated the protective function of ASA in the cardiovascular system (Deharo et al. 2014; Mahaffey et al. 2013) and immune system (Javeed et al. 2011). Moreover, recent experiments in our laboratory demonstrated that ASA pre-treatment would provide significant protective effects to chicken myocardial cells in vivo, for instance, the heat stress-induced increases of myocardial cell damage-related enzymes such as CK-MB and LDH were significantly supressed, and the pathological changes of myocardial cells such as acute degeneration were also reduced (Wu et al. 2015). ASA used as a painkiller was initially considered to function via platelet interactions (Patrono et al. 2005). However, its potential cardiovascular protective effect has been increasingly more appreciated. Although nonsteroidal anti-inflammatory drugs (NSAIDs) also have anti-inflammatory and antiplatelet properties similar to ASA, few of them can exert the same myocardial protective effects against acute myocardial infarction (AMI) (Solomon et al. 2002). Therefore, ASA may confer protection to the heart by another mechanism. Based on the myocardial protective function of HspB1, the ability of ASA to increase HspB1 expression may be reasonably speculated as a basis for using this drug to protect the heart from heat stress-induced injury. Therefore, in this study, we focused on exploring the possible connection between the heat stress-related injury in chicken myocardial cells and ASA-induced HspB1 expression in vitro.
Materials and methods
Cell stress model
Primary cultured chicken myocardial cells were provided by the Shanghai Fu Meng Biological Technology Ltd (Shanghai, China). Cells were cultured (37 °C and 5 % CO2) on the petri dishes for 72 h until the fusion rate was higher than 90 %. The cell culture medium contained 20 % fetal bovine serum (FBS, SH30088.03, Hyclone, Logan, UT, USA), 100 IU penicillin, and streptomycin (SV30010, HyClone) in Dulbecco’s modified Eagle’s medium (NYH0954, Thermo Scientific, Waltham, MA, USA).
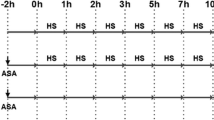
As shown in Fig. 1, primary cultured cells were divided into three groups: HS group (heat stress challenge), ASA-HS group (pre-treated with ASA before heat stress), and ASA group (treated with ASA alone). Except for cells in the HS group, those in the ASA-HS group and the ASA group were administered ASA 2 h before the heat stress phase. This time point was marked as −2 h. As soon as the heat stress phase began, myocardial cells in the ASA group were maintained under normal conditions and served as one of the control groups (non-heated cells); meanwhile, the other two groups, the HS group and the ASA-HS group, were exposed to heat stress by water bath (40 ± 1 °C) for different time span (0, 1, 2, 3, 5, 7, 10, 15, and 24 h). At each time point, the supernate of the cell culture media from each group was collected, and myocardial cells were harvested. Samples for biochemical analysis were frozen in liquid nitrogen. The final concentration of ASA in the cell culture medium was 1 mg/ml, which was determined by MTT assays (raw data was not shown in this paper).

Depiction of the heat-stressed model. Primary cultured myocardial cells were pre-treated with ASA or vehicle 2 h in prior (−2 h). As soon as the heat stress phase began, myocardial cells of the HS group and ASA-HS group were exposed to heat stress over a series of time points (0, 1, 2, 3, 5, 7, 10, 15, and 24 h). Samples were collected at the each time points in each group. ASA, administration with ASA at 1 mg/ml; HS, heat stress exposure by water bath 40 ± 1 °C; vehicle, same dosage of vehicle was used as a control; none, no specific treatment was used in the incubation
Additional cells were also cultured on poly-L-lysine (PLL) treated glass coverslips (product ID: 1014318, Sigma) for pathological and immunofluorescent analyses. Treatments and measurements were the same as those mentioned above.
Detection of heart damage-related enzymes
Supernatants of cultured cells after different treatments were collected for enzyme testing. The enzyme activities of lactate dehydrogenase (LDH) and creatine kinase-myocardial band (CK-MB) in all samples were measured according to the instructions given in the commercial kits (Nanjing Jiancheng Biochemical Reagent Co. Ltd., Nanjing, China) by using a clinical biochemical indicator auto-analyzer (Vital Scientific NV, Dieren, The Netherlands). Each sample was analyzed three consecutive times.
Cytopathological observations
For cytopathological tests, myocardial cells cultured on PLL-pre-treated coverslips in vitro were fixed in paraformaldehyde overnight and were washed with PBS (pH 7.4) twice before staining. All sections were stained with hematoxylin and eosin (H&E), and the images were obtained using a light microscope.
Detection of HspB1 in myocardial cells in vitro by ELISA assays
Total proteins from primary cultured myocardial cells were extracted by using the Protein Extraction Reagent (WB-0071, Dingguo Changsheng Biotechnology Co., Ltd., Nanjing, China). All protein concentrations were measured using a Micro BCA assay kit (23235, Thermo Scientific). HspB1 concentrations in all three groups were measured by using the Chicken Heat Shock Protein 27 (HSPB1) ELISA Kit (MBS700383, MyBioSource, San Diego, CA, USA) according to the instructions given in the commercial kits.
Immunofluorescence analysis
After being fixed with paraformaldehyde and washed with PBS (0.01 M, pH 7.4) twice, cells cultured on coverslips were transferred into PBS solution (0.1 M, pH 7.4) containing 0.5 % Triton x-100 for 15 min. Non-specific antigens were blocked by 5 % BSA for 30 min at room temperature. Thereafter, the HspB1 specific monoclonal antibody (ab49919, Abcam) was added at 1:200 dilution (4 °C, overnight). After incubation with 100 μL of goat anti-mouse IgG-FITC (BA1101, BOSTER, China) secondary antibody at a 1:50 dilution for 1 h at 37 °C, nuclei were stained with 50 μL DAPI for 15 min. The slides were mounted with anti-quenching mounting reagent (AR1109, BOSTER, Wuhan, China) and observed with a fluorescence microscope (Cx41-32rfl, Olympus, Japan).
In order to obtain optimal images for further semi-quantitative analysis, the exposure time of the fluorescence microscope was adjusted from 200 ms to 800 ms. Differences in average optical density (AOD) between the nucleus and the cytoplasm were analyzed using Image-Pro Plus 6.0 software. Five views were captured and analyzed in each coverslip.
Statistical analysis
The professional software Curve Expert 1.3 was used to generate standard curves for ELISAs. Data were compared with the baseline level (0 h in the HS group) by one-way analysis of variance (ANOVA) followed by Fisher’s least significant difference (LSD) test using the Statistical Package for Social Sciences (SPSS version 20.0 for Windows). Significant differences were indicated by P < 0.05, and highly significant differences were indicated by P < 0.01 in this study. Duncan’s multiple range test was used to analyze differences in HspB1 levels among three groups at each time point. All raw data presented were expressed as the mean ± standard deviation (SD). Differences in AODs between the nucleus and cytoplasm were analyzed by t test. All experiments were repeated three times.
Results
Variations of myocardial cell damage-related enzymes in vitro
Variations of CK-MB and LDH activities in the supernatant of myocardial cells in vitro are displayed in Fig. 2. Except for the return at 5 and 24 h of heat stress, the levels of CK-MB activities in the HS group significantly increased (P < 0.05) from 2 h of heat stress until the end of the experiment (0.50 ∼ 0.8-fold higher than baseline level). The levels of LDH increased significantly at the time points of 1, 2, 3, 5, and 15 h, with the highest level (3101 ± 185.9 IU/L) at 5 h of heat stress in the HS group. With ASA pre-treatment in vitro, levels of CK-MB slightly increased only at 2 h (0.45-fold higher), 3 h (0.54-fold higher), and 15 h (0.43-fold higher) of heat stress, while LDH levels increased only at 5 h (0.53-fold higher) of heat stress in the ASA-HS group. No significant changes of CK-MB and LDH were detected in the ASA group.

Variations of CK-MB and LDH in supernatant of cell culture media in vitro. CK-MB and LDH levels in each group were compared with the baseline level (0 h in the HS group, as well as at −2 h in ASA-HS and ASA groups). *P < 0.05; **P < 0.01
Cytopathological changes
Cytopathological changes of the primary cultured chicken myocardial cells after heat treatment are shown in Fig. 3. In the HS group, obvious acute vacuolar degeneration was observed in the cytoplasm of myocardial cells immediately upon exposure to high temperature for 1 h. This vacuolar degeneration, accompanied by granular degeneration, developed as the period of heat stress was prolonged and finally led to karyopyknosis and inadherence, since 15 h of heat exposure. In the ASA-HS group, vacuolar and granular degeneration could be observed in the cytoplasm of the myocardial cells after 1 h of heat stress exposure, but the degeneration was slighter than that in the HS group. Although such degeneration still could be observed until 15 h of heat stress, it lessened in the later period. Except for the degeneration, no further obvious pathological lesions were observed in the myocardial cells of the ASA-HS group. Without heat stress involved, no obvious pathological change was observed in the ASA group from the beginning to the end of the experiment.

Representative cytopathological images of chicken myocardial cells following heat exposure. Hematoxylin and eosin staining. Scale bar = 10 μm. HS group: No obvious pathological change was found before heat stress exposure (0 h). However, since being exposed for 1 h, myocardial cells displayed acute vacuolar degeneration, and such injury developed into karyopyknosis and inadherence since 15 h heat exposure. ASA-HS group: Degeneration can be observed since 1 h heat exposure and maintained until 15 h. But the degeneration was obviously slighter than that in the HS group. ASA group: no significant pathology change can be observed from the beginning to the end
Variations of HspB1 expression in cultured myocardial cells
Variations of HspB1 expression in cultured myocardial cells are displayed in Table 1. In the HS group, significant induction of HspB1 (P < 0.05) was detected at 5 and 15 h of heat stress, compared to that at 0 h of heat stress. However, induction of HspB1 levels in the cultured myocardial cells was continuous from 2 to 5 h, as well as from 10 to 24 h of heat stress, both in the ASA group (P < 0.05 at least) and the ASA-HS group (P < 0.01). Statistically, levels of HspB1 induced by ASA pre-treatment were much more remarkable in the myocardial cells than that by heat stress. Average HspB1 expression levels in the myocardial cells in the ASA-HS and ASA groups were 45.62-fold and 38.54-fold higher than that in the HS group, respectively.
Immunofluorescence detection of HspB1 in cultured myocardial cells
Immunofluorescence assays were performed to determine localization of HspB1 in the cultured myocardial cells (Fig. 4). The fluorescent signals were detected both in the cytoplasm and the nucleus of the cultured myocardial cells before heat exposure in the HS group. As the heat stress was prolonged, significantly stronger (P < 0.01) positive signals were located in the nucleus of myocardial cells in the HS and ASA-HS groups. However, positive HspB1 signals with similar intensity were located both in the cytoplasm and nucleus of myocardial cells from the beginning to the end of the experiment in the ASA group.

Distribution of HspB1 in cultured myocardial cells by immunofluorescence staining. Heat stress caused HspB1 relocalization into the nucleus in the HS and ASA-HS groups but not in the ASA group. Differences in average optical densities (AODs) between the nucleus and cytoplasm were analyzed by t test. *P < 0.05; **P < 0.01; scale bar = 10 μm; HspB1 (green color, FITC); nuclei (blue color, DAPI)
Discussion
In this study, ASA presented HspB1 inducing function as well as heat stress resistance in vitro as expected. Heat stress caused injury in myocardial cells was evidenced by the morphological lesions and cardiomyocyte damage-related enzyme activities. As reported, upon exposure to heat stress, the permeability of the myocardial cell membrane will increase and lead to extracellular release of a series of enzymes, including CK-MB and LDH. These enzymes are generally designated as cardiomyocyte damage-related enzymes and further regarded as important indicators for judging acute myocardial injury (Amani et al. 2013; Chen et al. 2013; Chon et al. 2013; Wu et al. 2013), especially CK-MB (Zeren et al. 2013). In addition to these enzymes, cytopathological changes like swollen in size, uneven staining in the cytoplasm and vacuolar degeneration were clearly identified in a lot of researches (Chen et al. 2015; Tang et al. 2013). According to these myocardial cell damage markers, it is clearly to find that overexpressed HspB1 by ASA was accompanied with relatively slight heat stress injury in myocardial cells, while severe cell damage appeared when ASA was not involved and the HspB1 level was low. These facts suggested that ASA exerted valuable anti-heat stress activity in myocardial cells in vitro, and inducing abundant HspB1 can be closely associated with this function. Meanwhile, even though high temperature itself also increased the expression of HspB1 at a few time points, the serious myocardial cell damage caused by heat stress challenge without ASA administration revealed that the HspB1 induced by high temperature alone was obviously insufficient to compensate for the massive consumption under stress circumstance. Generally, ASA was capable of reducing the heat stress injury in cultured myocardial cells and inducing sufficient HspB1 may be one of the key elements in anti-heat stress injury.
Another important characteristic of ASA that would contribute to its potential use as an anti-heat stress medicine was the safety profile. When the myocardial cells were treated with ASA but not exposed to heat stress, they showed concentration levels of HspB1 that were as high as those in the ASA-HS group. However, the morphological observation and the detection of cardiomyocyte damage-related enzymes indicated no significant pathological changes in the ASA group. This fact indicated that ASA acted as HspB1 inducer but no detectable insult was caused. Pre-treatments with many physical, chemical, and biological stress factors were reported to be able to cause an increase of HspB1 (Lindquist 1986). However, most of them also led to irreversible injury, which actually sometimes even more serious than that with heat stress. Some mineral salts, such as sodium arsenite, were found to induce Hsps efficiently but were quite toxic in animals with a 20 % mortality rate (Lappas et al. 1994; Ribeiro et al. 1994). The ability of ASA to induce HspB1 without leading to visible injury is extraordinarily valuable, although much more work is still needed for its development as a commercial anti-stress medicine.
The translocation of HspB1 after different treatments also revealed interesting information in this study. As long as the heat stress was involved in the experiment, HspB1 signals were localized stronger in the nucleus than in the cytoplasm of myocardial cells. Meanwhile, although HspB1 was overexpressed by ASA pre-treatment alone, in the absence of high-temperature exposure, positive HspB1 signals in the nucleus of myocardial cells presented no difference to that in the cytoplasm. This translocation phenomenon can be explained by the theory which has been partly described in many prior studies, but has it not been fully stated. As oligomers or dipolymers, HspB1 possesses dual protective functions against stress factors. It harbors the chaperone function only when in the form of large, unphosphorylated oligomers (Rogalla et al. 1999), and it was believed that non-phosphorylated HspB1 can stabilize cytoskeletal components, such as actin, via its chaperone function (Hollander et al. 2004). Under specific circumstances, these oligomeric HspB1 will dissociate into phosphorylated dipolymers. The phosphorylated form of HspB1 is a potential anti-apoptotic molecule that may directly interfere with cell death signaling pathways (Benn et al. 2002; de Graauw et al. 2005). It is localized within the perinuclear region at normal conditions (37 °C) and is relocated to the nucleus after heat stress (Arrigo et al. 1988). Stress from heat or ATP-depletion causes HspB1 to form large (∼106 kDa) detergent insoluble structures inside the nucleus (Arrigo et al. 1988; Arrigo 1994), which are visible as granules (Adhikari et al. 2004; Bryantsev et al. 2002; Loktionova et al. 1996) and also contain heat-denatured proteins. Therefore, the protective roles of HspB1 are also regulated by phosphorylation and translocation. Unlike high temperature which induced HspB1 and led to its translocation into the nucleus of myocardial cells, ASA pre-treatment induced HspB1 more efficiency without causing its translocation. These facts suggested that ASA activated the overexpression of HspB1 by another mechanism different from heat stress.
Both high-temperature exposure and low-dose ASA pre-treatment have been well described to be able to induce HspB1 (Amberger et al. 1999; Ebert et al. 2005; Sandoval-Montiel et al. 2013; Yan et al. 2009; Yu et al. 2009). Less notable, but no less meaningful, was the finding that instead of a stably variation, the increasing tendency of HspB1 fluctuated as the experiment proceeded under different treatments. This phenomenon reflected that the classic negative feedback regulation of the expression of Hsps also plays a functional role in HspB1, an ATP-independent sHsp (Faiella et al. 2012; Morimoto et al. 1992). As an important transcriptional activator, heat shock factor 1 (HSF-1) is occupied by Hsp70 under normal conditions (Knauf et al. 1996; Zuo et al. 1994). When exposed to stress factors, Hsp70 dissociates from HSF-1 and binds to degenerated proteins. Thereafter, HSF-1 oligomerizes into a trimer capable of transport into the nucleus. Once the trimeric HSF-1 is conjugated with heat shock element (HSE), the transcription of a series of Hsps is initiated. As the concentration of Hsps is elevated, HSF-1 again becomes competitively occupied by Hsps, especially Hsp70, and then transcription and expression of additional Hsps are suppressed, including HspB1.
Previously, we conducted a preliminary assessment of the protective capacity of ASA and the association with HspB1 in an animal model (Wu et al. 2015). In the extracorporal study, we demonstrated consistent results in cultured myocardial cells. The concurring in vivo and in vitro results indicated that heat stress leads to acute injury of chicken myocardial cells. Although no structural damage was observed in the pathological analysis, acute degeneration and high levels of karyopyknosis accompanying the changes of cardiomyocyte damage-related enzymes revealed serious myocardial dysfunction and metabolic disorders. These types of injuries can be lethal or at least involve impairment of productivity and lead to commercial loss in breeding industries (Kamboh et al. 2013; Ma et al. 2013; Zhao et al. 2013). Notably, the expression of HspB1 was more sensitive to ASA in the extracorporeal model. In vitro, with or without exposure to hyperthermia, levels of HspB1 in the myocardial cells in the two ASA-treated groups were almost two orders of magnitude higher than that in the HS group in which the cardiomyocytes were only stimulated with high temperature. Although the expression of HspB1 was also elevated in the animal models after ASA pre-treatments, the HspB1 expression in the heart tissue was not as sharp as that in the extracorporeal cultured cells. This finding suggested that a more complicated regulatory mechanism exists in vivo which would limit the overexpression of HspB1. At sufficient levels, HspB1 has been widely considered to serve multiple protective functions, but its overabundance may protect mutant cells from programmed death or even lead to cancer. A previous study associated stromal expression of HspB1 (Hsp27) with cancer development (Schweiger et al. 2015). Some researchers believe that inappropriate expression of Hsps, especially HspB1, can block apoptosis pathways and actively be involved in various processes such as tumor cell proliferation, invasion, metastases, and death (Lianos et al. 2015). The suppression of HspB1 suppression possibly serves as an important safeguard to prevent myocardial cells from tumorigenesis. Moreover, this regulatory system obviously involves multiple factors and organs since the isolated myocardial cell model presented relatively unlimited HspB1 expression in vitro. However, the precise mechanism requires further investigations.
References
Adhikari AS, Sridhar Rao K, Rangaraj N, Parnaik VK, Mohan Rao C (2004) Heat stress-induced localization of small heat shock proteins in mouse myoblasts: intranuclear lamin A/C speckles as target for alphaB-crystallin and Hsp25. Exp Cell Res 299:393–403. doi:10.1016/j.yexcr.2004.05.032
Amani M, Jeddi S, Ahmadiasl N, Usefzade N, Zaman J (2013) Effect of HEMADO on level of CK-MB and LDH enzymes after ischemia/reperfusion injury in isolated rat heart. Bio Impacts BI 3:101–104. doi:10.5681/bi.2013.003
Amberger A, Hala M, Saurwein-Teissl M, Metzler B, Grubeck-Loebenstein B, Xu Q, Wick G (1999) Suppressive effects of anti-inflammatory agents on human endothelial cell activation and induction of heat shock proteins. Mol Med 5:117–128
Arrigo APaL J (1994) Expression and function of the low-molecular-weight heat shock proteins. In: Morimoto RI, Tissieres A, Georgopoulos C (eds) The biology of heat shock proteins and molecular chaperones., pp 335–373
Arrigo AP, Suhan JP, Welch WJ (1988) Dynamic changes in the structure and intracellular locale of the mammalian low-molecular-weight heat shock protein. Mol Cell Biol 8:5059–5071
Benn SC et al (2002) Hsp27 upregulation and phosphorylation is required for injured sensory and motor neuron survival. Neuron 36:45–56
Bryantsev AL, Loktionova SA, Ilyinskaya OP, Tararak EM, Kampinga HH, Kabakov AE (2002) Distribution, phosphorylation, and activities of Hsp25 in heat-stressed H9c2 myoblasts: a functional link to cytoprotection. Cell Stress Chaperones 7:146–155
Chen TH, Yang YC, Wang JC, Wang JJ (2013) Curcumin treatment protects against renal ischemia and reperfusion injury-induced cardiac dysfunction and myocardial injury. Transplant Proc 45:3546–3549. doi:10.1016/j.transproceed.2013.09.006
Chen H, Adam A, Cheng Y, Tang S, Hartung J, Bao E (2015) Localization and expression of heat shock protein 70 with rat myocardial cell damage induced by heat stress in vitro and in vivo. Mol Med Rep 11:2276–2284. doi:10.3892/mmr.2014.2986
Chon H, Lee S, Yoon SY, Lee EK, Chang SI, Choo J (2013) SERS-based competitive immunoassay of troponin I and CK-MB markers for early diagnosis of acute myocardial infarction Chem Commun (Camb) doi:10.1039/c3cc47850e
de Graauw M, Tijdens I, Cramer R, Corless S, Timms JF, van de Water B (2005) Heat shock protein 27 is the major differentially phosphorylated protein involved in renal epithelial cellular stress response and controls focal adhesion organization and apoptosis. J Biol Chem 280:29885–29898. doi:10.1074/jbc.M412708200
Deharo P et al. (2014) Fixed-dose aspirin-clopidogrel combination enhances compliance to aspirin after acute coronary syndrome Int J Cardiol doi:10.1016/j.ijcard.2013.12.194
Ebert MP et al (2005) Protective role of heat shock protein 27 in gastric mucosal injury. J Pathol 207:177–184. doi:10.1002/path.1815
Ellis FP (1972) Mortality from heat illness and heat-aggravated illness in the United States. Environ Res 5:1–58
Faiella L, Piaz FD, Bisio A, Tosco A, De Tommasi N (2012) A chemical proteomics approach reveals Hsp27 as a target for proapoptotic clerodane diterpenes. Mol Bio Syst 8:2637–2644. doi:10.1039/c2mb25171j
Gao Z et al (2013) Inhibition of heat-induced apoptosis in rat small intestine and IEC-6 cells through the AKT signaling pathway. BMC Vet Res 9:241. doi:10.1186/1746-6148-9-241
Hollander JM, Martin JL, Belke DD, Scott BT, Swanson E, Krishnamoorthy V, Dillmann WH (2004) Overexpression of wild-type heat shock protein 27 and a nonphosphorylatable heat shock protein 27 mutant protects against ischemia/reperfusion injury in a transgenic mouse model. Circulation 110:3544–3552. doi:10.1161/01.CIR.0000148825.99184.50
Javeed A, Hou Y, Duan K, Zhang B, Shen H, Cao Y, Zhao Y (2011) Aspirin significantly decreases the nonopsonic phagocytosis and immunogenicity of macrophages in mice. Inflamm Res 60:389–398. doi:10.1007/s00011-010-0283-4
Kamboh AA, Hang SQ, Bakhetgul M, Zhu WY (2013) Effects of genistein and hesperidin on biomarkers of heat stress in broilers under persistent summer stress. Poult Sci 92:2411–2418. doi:10.3382/ps.2012-02960
Kato K, Hasegawa K, Goto S, Inaguma Y (1994) Dissociation as a result of phosphorylation of an aggregated form of the small stress protein, hsp27. J Biol Chem 269:11274–11278
Knauf U, Newton EM, Kyriakis J, Kingston RE (1996) Repression of human heat shock factor 1 activity at control temperature by phosphorylation. Genes Dev 10:2782–2793
Lambert H, Charette SJ, Bernier AF, Guimond A, Landry J (1999) HSP27 multimerization mediated by phosphorylation-sensitive intermolecular interactions at the amino terminus. J Biol Chem 274:9378–9385
Lappas GD, Karl IE, Hotchkiss RS (1994) Effect of ethanol and sodium arsenite on HSP-72 formation and on survival in a murine endotoxin model. Shock 2:34–39, discussion 40
Lianos GD, Alexiou GA, Mangano A, Rausei S, Boni L, Dionigi G, Roukos DH (2015) The role of heat shock proteins in cancer. Cancer Lett 360:114–118. doi:10.1016/j.canlet.2015.02.026
Lindquist S (1986) The heat-shock response. Annu Rev Biochem 55:1151–1191. doi:10.1146/annurev.bi.55.070186.005443
Liu L et al (2007) Over-expression of heat shock protein 27 attenuates doxorubicin-induced cardiac dysfunction in mice. Eur J Heart Fail 9:762–769. doi:10.1016/j.ejheart.2007.03.007
Loktionova SA, Ilyinskaya OP, Gabai VL, Kabakov AE (1996) Distinct effects of heat shock and ATP depletion on distribution and isoform patterns of human Hsp27 in endothelial cells. FEBS Lett 392:100–104
Ma X et al. (2013) Heat shock protein 27 attenuates neointima formation and accelerates reendothelialization after arterial injury and stent implantation: importance of vascular endothelial growth factor up-regulation FASEB journal: official publication of the Federation of American Societies for Experimental Biology doi:10.1096/fj.13-230417
Mahaffey KW et al. (2013) Association of aspirin dose and vorapaxar safety and efficacy in patients with non-ST-segment elevation acute coronary syndrome (from the TRACER Trial) the American journal of cardiology doi:10.1016/j.amjcard.2013.11.052
Morimoto RI, Sarge KD, Abravaya K (1992) Transcriptional regulation of heat shock genes. A paradigm for inducible genomic responses. J Biol Chem 267:21987–21990
Patrono C, Garcia Rodriguez LA, Landolfi R, Baigent C (2005) Low-dose aspirin for the prevention of atherothrombosis. N Engl J Med 353:2373–2383. doi:10.1056/NEJMra052717
Piestun Y, Druyan S, Brake J, Yahav S (2013) Thermal manipulations during broiler incubation alter performance of broilers to 70 days of age. Poult Sci 92:1155–1163. doi:10.3382/ps.2012-02609
Rhoads ML, Kim JW, Collier RJ, Crooker BA, Boisclair YR, Baumgard LH, Rhoads RP (2010) Effects of heat stress and nutrition on lactating Holstein cows: II. Aspects of hepatic growth hormone responsiveness. J Dairy Sci 93:170–179. doi:10.3168/jds.2009-2469
Ribeiro SP, Villar J, Downey GP, Edelson JD, Slutsky AS (1994) Sodium arsenite induces heat shock protein-72 kDa expression in the lungs and protects rats against sepsis. Crit Care Med 22:922–929
Rogalla T et al (1999) Regulation of Hsp27 oligomerization, chaperone function, and protective activity against oxidative stress/tumor necrosis factor alpha by phosphorylation. J Biol Chem 274:18947–18956
Sandoval-Montiel AA, Zentella-de-Pina M, Ventura-Gallegos JL, Frias-Gonzalez S, Lopez-Macay A, Zentella-Dehesa A (2013) HSP-72 accelerated expression in mononuclear cells induced in vivo by acetyl salicylic acid can be reproduced in vitro when combined with H2O2. PLoS One 8:e65449. doi:10.1371/journal.pone.0065449
Saravanan G, Ponmurugan P, Sathiyavathi M, Vadivukkarasi S, Sengottuvelu S (2013) Cardioprotective activity of Amaranthus viridis Linn: effect on serum marker enzymes, cardiac troponin and antioxidant system in experimental myocardial infarcted rats. Int J Cardiol 165:494–498. doi:10.1016/j.ijcard.2011.09.005
Scheers-Masters JR, Schootman M, Thach BT (2004) Heat stress and sudden infant death syndrome incidence: a United States population epidemiologic study. Pediatrics 113:e586–592
Schweiger T et al (2015) Stromal expression of heat-shock protein 27 is associated with worse clinical outcome in patients with colorectal cancer lung metastases. PLoS One 10:e0120724. doi:10.1371/journal.pone.0120724
Solomon DH, Glynn RJ, Levin R, Avorn J (2002) Nonsteroidal anti-inflammatory drug use and acute myocardial infarction. Arch Intern Med 162:1099–1104
N. R. St-Pierre* BC, and G. Schnitkey† (2003) Economic losses from heat stress by US livestock industries. J Dairy Sci 86:E52–E77
Tang S et al (2013) Localization and expression of Hsp27 and alphaB-crystallin in rat primary myocardial cells during heat stress in vitro. PLoS One 8:e69066. doi:10.1371/journal.pone.0069066
Wu NC, Chen TH, Yang YC, Liao FT, Wang JC, Wang JJ (2013) N-acetylcysteine improves cardiac contractility and ameliorates myocardial injury in a rat model of lung ischemia and reperfusion injury. Transplant Proc 45:3550–3554. doi:10.1016/j.transproceed.2013.09.005
Wu D et al. (2015) Acetyl salicylic acid protected against heat stress damage in chicken myocardial cells and may associate with induced Hsp27 expression cell stress & chaperones doi:10.1007/s12192-015-0596-x
Yan J, Bao E, Yu J (2009) Heat shock protein 60 expression in heart, liver and kidney of broilers exposed to high temperature. Res Vet Sci 86:533–538. doi:10.1016/j.rvsc.2008.09.002
Yu J, Tang S, Bao E, Zhang M, Hao Q, Yue Z (2009) The effect of transportation on the expression of heat shock proteins and meat quality of M. longissimus dorsi in pigs. Meat Sci 83:474–478. doi:10.1016/j.meatsci.2009.06.028
Zeren G et al (2013) [Relation of heart-type fatty acid-binding protein with the degree and extent of atherosclerosis in patients with non-ST elevation acute coronary syndrome]. Turk Kardiyol Dern Ars 41:610–616. doi:10.5543/tkda.2013.26974
Zhao W, Wisniewski M, Wang W, Liu J, Liu Y (2013) Heat-induced oxidative injury contributes to inhibition of Botrytis cinerea spore germination and growth World journal of microbiology & biotechnology doi:10.1007/s11274-013-1513-z
Zuo J, Baler R, Dahl G, Voellmy R (1994) Activation of the DNA-binding ability of human heat shock transcription factor 1 may involve the transition from an intramolecular to an intermolecular triple-stranded coiled-coil structure. Mol Cell Biol 14:7557–7568
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (31372403), the Natural Science Foundation of the Jiangsu Province (Grant No. BK20140107), the Postgraduate Student Research and Innovation Project of Jiangsu Province (KYLX15_0558), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the Sino-German Agricultural Cooperation Project of the Federal Ministry of Food, the Agriculture and Consumer Production, Berlin, Germany.
Author information
Authors and Affiliations
Corresponding author
Additional information
Di Wu and Miao Zhang contributed equally to this work.
Rights and permissions
About this article

Cite this article
Wu, D., Zhang, M., Xu, J. et al. In vitro evaluation of aspirin-induced HspB1 against heat stress damage in chicken myocardial cells. Cell Stress and Chaperones 21, 405–413 (2016). https://doi.org/10.1007/s12192-016-0666-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12192-016-0666-8