
Abstract
Genotypes of 260 individuals with hemoglobin H (Hb H) disease originating from various provinces in southern Thailand were characterized by multiplex PCR (M-PCR) and reverse dot blot hybridization (RDB). M-PCR was used to amplify target fragments and then hybridized with allele-specific oligonucleotide (ASO) probes which were bound on a nylon membrane. A total of eight α-thalassemia (α-thal) mutations, which produced eight Hb H disease genotypes (α0-thal/α+-thal), were detected. The most common form of α0-thal was −SEA with a frequency of 99.23%. The other form (0.77%) of α0-thal mutation was a THAI deletion (−THAI). The deletional α+-thal mutations comprised 3.7 kb (-α3.7) and 4.2 kb (-α4.2) deletions which were found in 172 (66.15%) and 5 (1.92%) alleles, respectively. The incidence of non-deletional α+-thal in decreasing order was Hb Constant Spring (Hb CS, αCS) 28.85%, Hb Quong Sze (Hb QS, αQS) 1.54%, and Hb Paksé (Hb PS, αPS) 0.77%. The genotype characterization of Hb H disease and the development of the RDB technic for detection of α-thal mutations presented in this study enable the prenatal diagnosis of Hb Bart’s hydrops fetalis syndrome.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alpha thalassemia (α-thal) is caused by reduced or absent synthesis of α-globin chains. It has two clinically significant forms: hemoglobin Bart’s hydrops fetalis syndrome and Hb H disease. The Hb Bart’s hydrops fetalis is caused by deletion of all four α-globin genes, characterized by fetal onset of generalized edema, pleural and pericardial effusions, and severe hypochromic anemia. Death usually occurs in the neonatal period. Hb H disease is caused by the loss of function of three α-globin genes and often manifested clinically as thalassemia intermedia. The clinical severity of patients is variable, depending on the type of α-globin mutations [1, 2].
Hb H disease is found in many parts of the world, including Southeast Asia and southern China [3]. In Thailand, it is estimated that 7000 infants with Hb H disease are born annually, and that there are 420,000 patients with Hb H disease in this country [4]. Around 20–30% of Thai populations are a carrier of either α0-thal or α+-thal. Hb H disease is mostly caused by compound heterozygote of α0- and α+-thal alleles. The commonest α°-thal mutation is the South East Asian deletion (−SEA) which removes approximately 19.3 kb DNA of the α-globin gene cluster [5]. For α+-thal, the rightward deletion (-α3.7) is the most common, followed by the leftward deletion (-α4.2). Hb CS is the most common non-deletional α+-thal mutation [5, 6]. A person with Hb H disease has one of the α0-thal deletions, which is at high risk of having infants with Hb Barts hydrops fetalis syndrome [5]. Thus, the molecular diagnosis of Hb H disease is significant, not only for elucidating the molecular pathology of the condition, but also for providing prenatal diagnosis in families at risk of Hb Bart’s hydrops fetalis syndrome. In this study, we applied the M-PCR and RDB technics to identify the major point mutations and large deletions of Hb H disease simultaneously in a single procedure.
Materials and methods
Subjects
A total of 260 samples from individuals with Hb H disease were collected at Songklanagarind hospital, Prince of Songkla University. All subjects were diagnosed through hematological data and Hb analysis. Hematological data were obtained using an automated blood cell counter (Sysmex XN 3000; Sysmex, Japan). Hb analysis was performed by either high-performance liquid chromatography (HPLC, Variant™; Bio-Rad Laboratories, Hercules, CA, USA) or capillary zone electrophoresis (CE, Capillarys 2; Sebia, Lisses, France).This study was approved by the ethics committee of Songklanagarind Hospital (EC 57-005-04-6-2). The genomic DNA was extracted from the blood sample by genomic DNA Mini Kit (Geneaid, Taiwan). The yield and purity of the DNA were determined by gel electrophoresis and spectrophotometer.
Design of primers and probes
The sequence data of α-like globin gene were obtained from NG_000006.1 of GenBank database. The data of gene deletion breakpoints and point mutations on α-like globin gene were obtained from globin database [7].
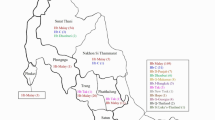
The M-PCR system included five sets of primers, which were designed to amplify the α-thal deletions of −SEA, −THAI, -α3.7 and -α4.2 types; point mutation regions of α2-globin gene [8,9,10,11,12,13,14,15]. The following primers were used: SEA1F, SEA2R, and A1B generated 389 bp and 249 bp fragments of mutant and normal alleles of −SEA, respectively; C3, C2, SEA1F, and A1B generated 460 bp and 249 bp fragments of −THAI and normal alleles, respectively; E and G generated 1762 fragments of -α4.2; A and Z were designed to amplify 1806 bp fragment of α2-globin gene for point mutation detection and characterization of -α3.7 subtypes (Fig. 1). The 1806 bp fragment included the mutation locations of Hb CS, Hb Suan Dok (Hb SD), Hb QS, and Hb PS (Fig. 1). The A and B primers generated the 1804 bp fragment of -α3.7 deletion, which was also used for characterization of -α3.7 subtypes (Fig. 1). ASO probes were designed both for deletion and point mutations. For large deletion, the mutant probes were designed across the junction regions, while normal probes were designed from normal sequences on normal PCR products. For point mutations, the ASO probes were designed to complementary with normal and mutant DNA sequences of each mutation [16, 17]. All probes were immobilized on a Biodyne C (Pall Biomedical, USA) nylon membrane strip through a covalently binding amino-modified ASO probes to the membrane-bound carboxyl group. The detailed information of the primers and probes is described in Tables 1 and 2. Some of the primers were biotinylated.

Schematic drawing of α-like globin gene cluster and the locations of amplification primers. The open boxes and the dark bars below the gene represent the locations of ASO probes for − α3.7 subtype characterization and the deletion sizes of α°-thal and α+-thal, respectively
Multiplex PCR and hybridization
The M-PCR reaction system was performed with a reaction volume of 50 µl containing 100 ng of genomic DNA, 3.5 µl of 10× reaction buffer, 10 µl of 5xQ solution (QIAGEN, Germany), 2.5 µl of DMSO, 5 µl of 2 mM dNTPs, 1.5 µl of 50 mM MgCl2, 1 µl of 30 µM each primers, 1 units of BIOTAQ™ DNA Polymerase (BIOLINE, USA) in a MJ Mini personal thermal cycler (Bio-Rad, USA) with preheating at 95 °C for 10 min, followed by 35 cycles of 95 °C for 1 min, 60 °C for 1 min, 72 °C for 1 min, and final extension at 72 °C for 10 min. Following amplification, the biotinylated PCR products were denatured and subjected to hybridization. Approximately 40–50 µl of amplified products were diluted with 500 µl of hybridization buffer (3xSSPE, 0.5% SDS) and denatured by boiling for 5 min. The hybridization reaction was carried out in 2 ml of hybridization buffer at 45 °C for 50 min. After hybridization, the membrane strip was washed in washing buffer (2xSSPE, 0.1% SDS) at 49 °C for 20 min and then incubated with streptavidin–alkaline phosphatase (Boehringer-Mannheim, Germany) in detection buffer (0.1 M tris pH 9.5, 0.1 M NaCl, 5 mM MgCl2) at room temperature for 45 min. The reaction was detected by transferring the membrane to enzymatic substrate solution, NBT and BCIP diluted in the detection buffer, and incubated in dark at room temperature until a blue–purple precipitate became visible at the hybrid probe. The reaction was stopped by rinsing with tap water. The results were interpreted by direct visualization (Fig. 2).

Example of reverse dot blot results and diagram of locations of oligonucleotide probes on a nylon membrane. The pictures show genotypes of normal individual and individuals with α-thalassemia. From top to bottom, the left column shows data from hybridization with the following genotypes αα/αα (a), αCSα/αα (b), −SEA/αCSα (c), −SEA/αPSα (d), and −SEA/αQSα (e); the right column shows data from hybridization with −SEA/-α3.7I (f), −SEA/-α3.7 II (g), −SEA/-α3.7 III (h), −SEA/-α4.2 (i), and −THAI/-α3.7I (j). The locations of hybridization probes complementary to 10 Hb H genotypes show in the diagram (k). The normal probes are listed at the top and the mutant probes are at the bottom. SEA-N is the common normal probe for −SEA and −THAI mutants; CS-N is the common normal probe for Hb CS, Hb PS, and -α4.2 mutants. 3.7-1, 3.7-2, 3.7-3, 3.7-4, 3.7-5, and 3.7-6 are the locations of hybridization probes for -α3.7 subtype characterization. No. denotes the sample labeled number
Results
Table 3 shows the frequencies of various α-globin genotypes in 260 patients with Hb H disease. All the cases were caused by the compound heterozygote of α°-thal allele combined with α+-thal allele (α0-thal/α+-thal). 178 (68.46%) had coinheritance of α°-thal with either α+-thal rightward deletion (-α3.7, n = 172) or α+ -thal leftward deletion (-α4.2, n = 5). 82 (31.54%) were due to compound heterozygosity of α°-thal with a non-deletional α-thal gene on the other chromosome. The most common type of α°-thal was −SEA with a frequency of 99.23%. The −THAI type of α-thal mutation was detected in the remaining patients (2 cases). The α+-thal alleles included -α3.7, -α4.2, -αCS, αQS, and αPS. The most common type was -α3.7, followed by αCS. The incidence of non-deletional α-thal in decreasing order was Hb CS (n = 75), Hb QS (n = 4), and Hb PS (n = 2). Three different subtypes of -α3.7 were found: subtype I (146, 84.88%), subtype II (20, 11.63%), and subtype III (6, 3.84%).
Discussion
In this study, the genotypes of 260 patients with Hb H disease from southern Thailand were characterized. The results are complementary to those already published from Thailand [18,19,20]. 178 patients (68.46%) have deletional genotype. The most common deletional genotype is −SEA/-α3.7 found in 172 patients (66.15%), followed by −SEA/-α4.2 5 patients (1.92%), and −THAI/-α3.7 1 patients (0.38%). Three subtypes of -α3.7 were found, subtypes I, II, and III, the most common is a subtype I, accounted for 84.88% of all -α3.7 types. Among these three subtypes, the values of erythrocyte indexes (mean corpuscular volume and mean corpuscular hemoglobin) were compared and found no significant differences (data not shown). 82 patients (31.54%) had non-deletional Hb H disease. The most prevalent genotype in this group was −SEA/αCS found in 74 patients (28.46%), followed by –SEA/-αQS 4 patients (1.54%) and –SEA/-αPS two patients (0.77%). Hb CS is the most common non-deletional α-globin mutation associated with Hb H disease, which the incidence varies between 1 and 8% throughout Thailand [21]. Hb CS and Hb PS are caused by mutation in the termination codon of the α2-globin gene [TAA > CAA (Gln) and TAA > TAT (Tyr), respectively]; resulting in reduced stability and abundance of the elongated α-globin variant [22]. The other unstable α-chain variant found by this study was Hb QS. Hb QS is a common non-deletional mutation in southern China that results from a nucleotide T-to-C substitution at codon 125 of α2-globin gene (CTG > CCG, Leu > Pro) [23]. Another unstable α-chain variant found in Thai population is Hb SD [24]. Hb SD is caused by a missense mutation at codon 109 of α2-globin gene (CTG > CGG, Leu > Arg), inducing Hb H disease in association with a −SEA allele. However, because the mutation was uncommon, there was no patient with this mutation in the present study.
The diagnosis of Hb H disease at the DNA level is important with respect to genetic counseling and identification of couples at risk for having pregnancies affected with Hb Bart’s hydrops fetalis syndrome or Hb H disease. Currently, however, the DNA diagnosis of Hb H disease needs several tests, which the separated tests are required for the deletion and non-deletion mutations [11, 25,26,27]. Both procedures are not practical and suitable for rapid diagnosis. In this study, we have developed a reverse dot blot hybridization technic for identification 8 common α-thal mutations in Thailand, which include two types of α0-thal: −SEA deletion and −THAI deletion, and 6 types of α+-thal, including two types of the deletion: 3.7 and 4.2 kb, and four types of point mutation: Hb CS, Hb PS, Hb QS, and Hb SD. The assay was based on a one-tube M-PCR for specific amplification of the α1- and α2-globin genes, as well as the common single- and double-gene deletions, followed by hybridization of the biotinylated amplification products to ASO probes bound to nylon membrane strips. The results demonstrated that the technic could identify all the major genotypes of Hb H disease (Table 3). Except in the case of 2 bp deletion at the polyA signal which could not be identified by this technic was further analyzed by direct DNA sequencing. The method as presented in this study provided a rapid tool for identifying the Hb H genotype and enabled screening of a sample for a large number of potential mutation sites [9]. It takes only 5–6 h for the results to become available, which can be easily performed with common equipment and has a low cost of less than US$10. In addition, the method was non-radioactive and not required specific technical skills.
References
Origa R, Moi P. (2016) Alpha-Thalassemia. GeneReviews® [Internet]. https://www.ncbi.nlm.nih.gov/books/NBK1435/ (cited 2017 Oct 20).
Chui DHK. Thalassemia. Hb H disease and Hb Bart’s hydrops fetalis. Ann N Y Acad Sci. 2005;1054:25–32.
Xu XM, Zhou YQ, Luo GX, Liao C, Zhou M, Chen PY, et al. The prevalence and spectrum of alpha and beta thalassaemia in Guangdong province: implications for the future health burden and population screening. J Clin Pathol. 2004;57:517–22.
Fucharoen S, Winichagoon P. Haemoglobinopathies in Southeast Asia. Indian J Med Res. 2011;134:498–506.
Chui DHK, Fucharoen S, Chan V. Hemoglobin H disease: not necessarily a benign disorder. Blood. 2003;101:791–800.
Pharephan S, Sirivatanapa P, Makonkawkeyoon S, Tuntiwechapikul W, Makonkawkeyoon L. Prevalence of α-thalassaemia genotypes in pregnant women in northern Thailand. Indian J Med Res. 2016;143:315–22.
Giardine B, van Baal S, Kaimakis P, Riemer C, Miller W, Samara M, et al. HbVar database of human hemoglobin variants and thalassemia mutations: 2007 update. Hum Mutat. 2007;28:206.
Lin M, Zhu JJ, Wang Q, Xie LX, Lu M, Wang JL, et al. Development and evaluation of a reverse dot blot assay for the simultaneous detection of common alpha and beta thalassemia in Chinese. Blood Cells Mol Dis. 2012;48:86–90.
Chan V, Yam I, Chen FE, Chan TK. A reverse dot-blot method for rapid detection of non-deletion alpha thalassaemia. Br J Haematol. 1999;104:513–5.
Ye BC1, Zhang Z, Lei Z. Oligonucleotide array for detection of common severe determinants of alpha thalassemia. J Biotechnol. 2005;115:1–9.
Zesong L, Ruijun G, Wen Z. Rapid detection of deletional alpha-thalassemia by an oligonucleotide microarray. Am J Hematol. 2005;80:306–8.
Baysal E, Huisman THJ. Detection of common deletional α-thalassaemia-2 determinants by PCR. Am J Hematol. 1994;46:208–13.
Chong SS, Boehm CD, Higgs DR, Cutting GR. Single-tube multiplex-PCR screen for common deletional determinants of α-thalassemia. Blood. 2000;95:360–2.
Liu YT, Old JM, Fisher CA, Weatherall DJ, Clegg JB. Rapid detection of α-thalassaemia deletions andα-globin gene triplication by multiplex polymerase chain reactions. Br J Haematol. 2000;108:295–9.
Chen TP, Liu TC, Chang CS, Chang JG, Tsai HJ, Lin SF. PCR-based analysis of alpha-thalassemia in Southern Taiwan. Int J Hematol. 2002;75:277–80.
Foglietta E, Bianco I, Maggio A, Giambona A. Rapid detection of six common Mediterranean and three non-Mediterranean α-thalassemia point mutations by reverse dot blot. Am J Hematol. 2003;74:191–5.
Puehringer H, Najmabadi H, Law HY, Krugluger W, Viprakasit V, Pissard S, et al. Validation of a reverse-hybridization StripAssay for the simultaneous analysis of common alpha-thalassemia point mutations and deletions. Clin Chem Lab Med. 2007;45:605–10.
Winichagoon P, Fucharoen S, Wasi P. The molecular basis of alpha-thalassemia in Thailand. Southeast Asian J Trop Med Public Health. 1992;23(Suppl 2):7–13.
Lithanatudom P, Khampan P, Duncan RS, Svasti S, Fucharoen S, Kangwanpong D, et al. The prevalence of alpha-thalassemia amongst Tai and Mon-Khmer ethnic groups residing in northern Thailand: a population-based study. Hematology. 2016;21:480–5.
Laosombat V, Viprakasit V, Chotsampancharoen T, Wongchanchailert M, Khodchawan S, Chinchang W, et al. Clinical features and molecular analysis in Thai patients with Hb H disease. Ann Hematol. 2009;88:1185–92.
Pornprasert S, Punyamung M. Detection of compound heterozygous of Hb constant spring and Hb Q-Thailand by capillary electrophoresis and high performance liquid chromatography. Indian J Hematol Blood Transfus. 2015;31:229–32.
Charoenkwan P, Taweephon R, Sae-Tung R, Thanarattanakorn P, Sanguansermsri T. Molecular and clinical features of Hb H disease in northern Thailand. Hemoglobin. 2005;29:133–40.
Yang Y, Lou JW, Liu YH, He Y, Li DZ. Screening and diagnosis of Hb Quong Sze [HBA2: c.377T> C (or HBA1)] in a prenatal control program for thalassemia. Hemoglobin. 2014;38:158–60.
Sanguansermsri T, Matragoon S, Changloah L, Flatz G. Hemoglobin Suan-Dok (α2 109(G16)LEU-ARGβ2). an unstable variant associated with α-thalassemia. Hemoglobin. 1979;3:161.
Bang-Ce Y, Hongqiong L, Zhuanfong Z, et al. Simultaneous detection of alpha-thalassemia and beta-thalassemia by oligonucleotide microarray. Haematologica. 2004;89:1010–2.
Old J, Henderson S. Molecular diagnostics for haemoglobinopathies. Expert Opin Med Diagn. 2010;4:225–40.
Harteveld CL, Kleanthous M, Traeger-Synodinos J. Prenatal diagnosis of hemoglobin disorders: present and future strategies. Clin Biochem. 2009;42:1767–79.
Acknowledgements
The authors thank the staff of the Hematology and Thalassemia Laboratory of the Department of Pathology, Faculty of Medicine, Prince of Songkla University for the technical support. This work was supported by a Grant (REC 57-005-04-6-2) from the Faculty of Medicine, Prince of Songkla University. The sponsors of this study are public or nonprofit organizations that support science in general.
Author information
Authors and Affiliations
Contributions
KN performed the research, analyzed the data, and wrote the manuscript. CN designed the research study, interpreted the data, and edited the manuscript. Final version of the article was read and approved by all of the authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interests regarding the publication of this paper.
About this article

Cite this article
Nittayaboon, K., Nopparatana, C. Molecular characterization of Hb H disease in southern Thailand. Int J Hematol 108, 384–389 (2018). https://doi.org/10.1007/s12185-018-2494-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-018-2494-3