
Abstract
Idiopathic aplastic anemia (AA) is a rare hematological complication of Down syndrome (DS). The safety and efficacy of immunosuppressive therapy (IST) in individuals with DS remain unknown. We used a standard regimen of IST, comprising antithymocyte globulin and cyclosporine A, to treat three children with DS and idiopathic acquired AA. Two patients achieved a hematological (complete or partial) response and became transfusion independent at the final follow-up. The third patient failed to respond to IST and underwent bone marrow transplantation from a human leukocyte antigen (HLA)-mismatched unrelated donor. None of the patients experienced severe or unexpected adverse events during IST. Our experience suggests that IST is a safe and reasonable treatment, even in individuals with DS who suffer from AA and lack an HLA-matched sibling donor.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Down syndrome (DS), which is the most frequent congenital chromosomal abnormality, is characterized by constitutional trisomy 21. It is associated with a predisposition to various hematological diseases, including transient abnormal myelopoiesis, myelodysplastic syndrome, and acute leukemia [1]. However, idiopathic acquired aplastic anemia (AA) is quite rare in individuals with DS, and only a few case reports are available [2–9]. Moreover, to our knowledge, there are no reports on the use of immunosuppressive therapy (IST) to treat patients with DS and AA. The safety and efficacy of IST in individuals with DS are therefore still unknown.
Here, we report three children with DS (two boys and one girl) who developed very severe or moderate AA and lacked a human leukocyte antigen (HLA)-matched sibling donor. They were treated with a standard IST regimen comprising antithymocyte globulin (ATG) and cyclosporine A (CSA). This is the first report that describes the clinical use of IST for AA patients with DS.
Case descriptions
From 1998 to 2013, two boys and one girl with DS who were diagnosed with idiopathic AA received IST comprising horse- (n = 1) or rabbit- (n = 2) derived ATG and CSA in our institutions. Information concerning their clinical findings and laboratory data was collected using a questionnaire. The institutional review board of Nagoya University Graduate School of Medicine approved this study. Written informed consent had been obtained from patients’ guardians at each institution.
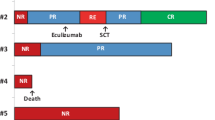
Patient characteristics and the clinical courses of each patient are summarized in Tables 1 and 2, respectively.
Case 1
Patient 1 was a 2-year-old boy with atrial septal defect and mild pulmonary hypertension. His family history was unremarkable. He presented with fever and his blood test showed marked pancytopenia, which fulfilled the criteria for very severe AA. His bone marrow smear revealed extreme hypocellularity without blast proliferation or dysplastic features.
He was diagnosed with AA, which did not respond to two courses of high-dose glucocorticoid therapy. On day 84 after initial diagnosis, he received IST, which comprised horse-derived ATG (15 mg/kg/day) on days 1–5 and CSA from day 1. CSA was started with the dose of 6 mg/kg/day orally, and then adjusted to maintain trough blood levels between 100 and 200 ng/mL. Although sufficient hematopoietic recovery was not achieved at 3 and 6 months, a complete response was seen 10 months after the start of IST. Twenty-eight months after IST, he developed immune thrombocytopenia, which was successfully treated with prednisolone. No other obvious adverse event was documented. CSA was gradually tapered and completely discontinued after 53 months. At the final follow-up (182 months after the start of IST), his performance status (PS) was excellent and blood cell counts were normal.
Case 2
Patient 2 was an 8-year-old girl with a medical history of anal atresia and hyperthyroidism that did not require any medication at the time of admission. Her family history was unremarkable. She presented with a tendency to bleed, and her blood test showed pancytopenia, with particularly prominent neutropenia, fulfilling the criteria for very severe AA. Her bone marrow was extremely hypocellular without blast proliferation or dysplasia, confirming the diagnosis of AA.
On day 39 after initial diagnosis, she received IST comprising rabbit-derived ATG (3.75 mg/kg/day) on days 1–5, CSA (administered in the same way as Case 1) from day 1, and granulocyte-colony stimulating factor (filgrastim, 5 µg/kg/day) for 56 days from day −12. Although sufficient hematopoietic recovery was not achieved at 3 and 6 months as observed in Case 1, a partial response occurred 11 months after IST. No adverse event was documented during IST, except for asymptomatic Epstein–Barr virus reactivation occurred 4 weeks after IST, which resolved immediately after the administration of rituximab, an anti-CD20 monoclonal antibody. At the final follow-up (52 months after the start of IST), she continued to receive orally administration of CSA. Her PS was excellent, with normal blood cell counts except for slight neutropenia (approximately 1.0 × 109/L).
Case 3
Patient 3 was an 8-year-old boy with ventricular septal defect and patent ductus arteriosus. His family history was unremarkable. He had no symptoms, and thrombocytopenia was noticed by chance. His initial blood test showed pancytopenia, fulfilling the criteria for moderate AA, and the bone marrow was hypocellular without blast proliferation.
He was diagnosed with AA, and an initial treatment with oral CSA combined with danazol was ineffective. Gradually, he developed a tendency to bleed, requiring periodic platelet transfusion. On day 278 after diagnosis, he received IST comprising rabbit-derived ATG (3.75 mg/kg/day) on days 1–5 and CSA (trough blood levels, 100–200 ng/mL). No obvious adverse event was documented during IST. Although at one point he became transfusion-independent, hematopoietic recovery was transient, so that he required both red blood cell and platelet transfusions. Eventually, his disease did not respond to IST, and he underwent allogeneic bone marrow transplantation (BMT) from an HLA-1/8-mismatched (HLA-C, determined using high-resolution DNA typing), unrelated donor 12 months after IST. We used a reduced intensity conditioning regimen comprising fludarabine (100 mg/m2) on days −6 to −3, cyclophosphamide (100 mg/kg) on days −6 to −3, rabbit-derived ATG (5 mg/kg) on days −6 to −3, and 2 Gy of total body irradiation on day −7. Graft-versus-host disease (GvHD) prophylaxis was administered with tacrolimus (0.02 mg/kg, continuous infusion) starting from day −1 and methotrexate at 15 mg/m2 on day 1 and 10 mg/m2 on days 3, 6, and 11. Although sustained engraftment was successfully obtained, at the final follow-up, he suffered from severe chronic GvHD with skin and lung involvement.
Discussion
Children with DS are predisposed to a broad spectrum of hematological abnormalities, including transient abnormal myelopoiesis, myelodysplastic syndrome, acute leukemia, and transient abnormal blood cell counts in the newborn [1]. In particular, the standardized incidence of leukemia in individuals with DS is reports to be 17.63 times higher compared with that of individuals without DS [10]. However, there are reports of only nine patients with idiopathic AA with DS (Supplementary Table 1) [2–9]. We are unaware of other reports describing the use of IST for treating AA in individuals with DS.
Two of our three patients showed a hematological response to IST within a year, although an obvious short-term response was not observed for at least the first 6 months. These two patients did not relapse, and their PS was excellent at the final follow-up. However, in the third patient there was a long delay between diagnosis and initiation of IST (278 days), and he did not respond to the treatment. This delay represents one of the several risk factors for a poor response to IST [11, 12].
First-line therapy for severe AA is allogeneic hematopoietic stem cell transplantation (HSCT) from an HLA-matched sibling donor, and patients who fail to respond to IST are rescued by the subsequent HSCT from an alternative donor, particularly an HLA-matched unrelated donor. However, the clinical experience with allogeneic HSCT for DS cases is limited, and the reported outcomes are significantly inferior compared to those for non-DS cases [8, 13]. For example, two of the three reported patients with DS and AA who received allogeneic BMT died of transplant-related complications [3, 8]. One of our patients who failed to respond to IST also underwent allogeneic BMT from an HLA-mismatched unrelated donor and suffered various complications, particularly severe chronic GvHD with lung involvement and adverse effects of systemic glucocorticoid therapy.
Children with DS are likely to have comorbidities, such as congenital heart disease, digestive tract anomalies, and immune dysfunction [14]. Sometimes they have drug sensitivities that differ from those of general population [15, 16]. Therefore, in addition to efficacy, tolerance to treatment is another important issue. Our patients did not experience severe or unexpected adverse effects from IST. Although asymptomatic Epstein–Barr virus reactivation occurred in one patient, it was completely controlled by the administration of rituximab. Moreover, during long-term follow-up, our patients did not experience clonal evolution, progression to leukemia, or development of paroxysmal nocturnal hemoglobinuria.
In summary, this is the first report to describe the clinical use of IST for treating patients with DS and AA. Favorable responses were achieved with acceptable adverse events in two of three cases. These findings suggest that IST is a safe and reasonable treatment even for individuals with DS who suffer from AA. Further investigation is warranted to confirm our observation.
References
Bruwier A, Chantrain CF. Hematological disorders and leukemia in children with Down syndrome. Eur J Pediatr. 2012;171:1301–7.
Erdogan G, Aksoy M, Dincol K. A case of idiopathic aplastic anaemia associated with trisomy-21 and partial endoreduplication. Acta Haematol. 1967;37:137–42.
Furuya A, Ishida M, Hodohara K, Yoshii M, Okuno H, Horinouchi A, et al. Epstein-Barr virus-related post-transplant lymphoproliferative disorder occurring after bone marrow transplantation for aplastic anemia in Down’s syndrome. Int J Clin Exp Pathol. 2014;7:438–42.
Gathwala G, Dalal P, Dalal JS, Choudhry O. Transient aplastic anemia in Down’s syndrome—a rare association. Eur J Med Genet. 2011;54:341–2.
Hanukoglu A, Meytes D, Fried A, Rosen N, Shacked N. Fatal aplastic anemia in a child with Down’s syndrome. Acta Paediatr Scand. 1987;76:539–43.
McWilliams NB, Dunn NL. Aplastic anemia and Down’s syndrome. Pediatrics. 1982;69:501–2.
Pavithran K, Raji NL. Aplastic anemia in Down’s syndrome. Am J Hematol. 2003;73:213.
Rubin CM, Mick R, Johnson FL. Bone marrow transplantation for the treatment of haematological disorders in Down’s syndrome: toxicity and outcome. Bone Marrow Transplant. 1996;18:533–40.
Weinblatt ME, Higgins G, Ortega JA. Aplastic anemia in Down’s syndrome. Pediatrics. 1981;67:896–7.
Hasle H, Clemmensen IH, Mikkelsen M. Risks of leukaemia and solid tumours in individuals with Down’s syndrome. Lancet. 2000;355:165–9.
Jeong DC, Chung NG, Cho B, Zou Y, Ruan M, Takahashi Y, et al. Long-term outcome after immunosuppressive therapy with horse or rabbit antithymocyte globulin and cyclosporine for severe aplastic anemia in children. Haematologica. 2014;99:664–71.
Yoshida N, Yagasaki H, Hama A, Takahashi Y, Kosaka Y, Kobayashi R, et al. Predicting response to immunosuppressive therapy in childhood aplastic anemia. Haematologica. 2011;96:771–4.
Goto H, Kaneko T, Shioda Y, Kajiwara M, Sakashita K, Kitoh T, et al. Hematopoietic stem cell transplantation for patients with acute lymphoblastic leukemia and Down syndrome. Pediatr Blood Cancer. 2015;62:148–52.
Weijerman ME, de Winter JP. Clinical practice. The care of children with Down syndrome. Eur J Pediatr. 2010;169:1445–52.
Garre ML, Relling MV, Kalwinsky D, Dodge R, Crom WR, Abromowitch M, et al. Pharmacokinetics and toxicity of methotrexate in children with Down syndrome and acute lymphocytic leukemia. J Pediatr. 1987;111:606–12.
Taub JW, Ge Y. Down syndrome, drug metabolism and chromosome 21. Pediatr Blood Cancer. 2005;44:33–9.
Acknowledgments
The authors would like to thank all of the patients and their families. The authors would also like to thank Ms. Yoshie Miura, Ms. Yuko Imanishi, and Ms. Hiroe Namizaki for their valuable assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
S.K. was supported by a grant from Sanofi K.K. The other authors declare that they have no conflict of interest. A summary of relevant information will be published with the manuscript.
Electronic supplementary material
Below is the link to the electronic supplementary material.
About this article

Cite this article
Suzuki, K., Muramatsu, H., Okuno, Y. et al. Immunosuppressive therapy for patients with Down syndrome and idiopathic aplastic anemia. Int J Hematol 104, 130–133 (2016). https://doi.org/10.1007/s12185-016-1997-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-016-1997-z