
Abstract
Thrombopoietin (TPO) is the major physiological regulator of platelet production. TPO binds the TPO receptor, activates JAK and STAT pathways, thus stimulating megakaryocyte growth and platelet production. There is no “sensor” of the platelet count; rather TPO is produced in the liver at a constant rate and cleared by TPO receptors on platelets. TPO levels are inversely proportional to the rate of platelet production. Early recombinant TPO molecules were potent stimulators of platelet production and increased platelets in patients with immune thrombocytopenia, chemotherapy-induced thrombocytopenia, myelodysplastic syndromes and platelet apheresis donors. Neutralizing antibodies formed against one recombinant protein and ended their development. A second generation of TPO receptor agonists, romiplostim and eltrombopag, has been developed. Romiplostim is an IgG heavy chain into which four TPO agonist peptides have been inserted. Eltrombopag is an oral small molecule. These activate the TPO receptor by different mechanisms to increase megakaryocyte growth and platelet production. After administration of either to healthy volunteers, there is a delay of 5 days before the platelet count rises and subsequently reaches a peak after 12–14 days. Both have been highly effective in treating ITP and hepatitis C thrombocytopenia. Studies in a wide variety of other thrombocytopenic conditions are underway.
Similar content being viewed by others



Avoid common mistakes on your manuscript.
Introduction
Thrombopoietin (TPO) was the last major hematopoietic growth factor to be identified, purified and cloned. The concept of a “thrombopoietin” had first entered the medical literature in 1958 when Kelemen proposed that a hematopoietic growth factor existed that regulated platelet production just as erythropoietin-regulated red cell production [1]. However, despite many decades of work by many laboratories, thrombopoietin remained elusive until its purification almost simultaneously by five separate research groups in 1994 [2–6]. Since then much has been learned about the structure and function of thrombopoietin as well as its clinical use.
In 1994 two recombinant thrombopoietins, recombinant human thrombopoietin (rhTPO) and pegylated, recombinant, human megakaryocyte growth and development factor (PEG-rhMGDF), began clinical development [7]. Both were potent stimulators of megakaryocyte growth and platelet production in both animal models and human subjects. A number of clinical trials demonstrated improved platelet counts in a variety of clinical situations ranging from chemotherapy-induced thrombocytopenia [8] to immune thrombocytopenia (ITP) [9]. Unfortunately, antibodies developed against PEG-rhMGDF in some healthy volunteers who received this agent and these antibodies neutralized its activity but also cross-reacted with and neutralized endogenous thrombopoietin to produce a paradoxical thrombocytopenia [10]. This concern about their antigenicity led to the discontinuation of any further development of recombinant thrombopoietins.
Given the promising results of the initial clinical work with recombinant thrombopoietins, a number of second generation thrombopoietin molecules, referred to hereafter as TPO receptor agonists, have now been developed [11, 12]. These were also effective stimulators of megakaryocyte growth and platelet production. Two of them, romiplostim and eltrombopag, have been approved in many countries for the treatment of ITP.
This chapter will review the structure and basic biology of thrombopoietin as well as its early development. An additional focus will be on the newer TPO receptor agonists describing their structure, function, pharmacology, known and potential adverse effects, and a brief overview of their current clinical use.
Thrombopoietin structure
TPO is synthesized primarily in the liver as a 353 amino acid precursor protein with a molecular weight of 36 kDa [2, 4, 13]. Following removal of a 21 amino acid signal peptide, the remaining 332 amino acid protein undergoes glycosylation and produces a glycoprotein with a molecular weight of 95 kDa on SDS-PAGE [10] and 57.5 kDa by mass spectrometry [14]. This glycoprotein is then released from the liver into the circulation with no apparent intracellular storage in the liver.
TPO is a member of the four-helix-bundle cytokine superfamily and has several unusual properties (Fig. 1). First, it is much larger than most other hematopoietic growth factors, such as granulocyte colony stimulating factor (G-CSF) and erythropoietin (EPO). Second, it has two distinct domains: an EPO-like domain (residues 1–153) and a carbohydrate-rich domain (residues 154–332) separated by site of potential proteolytic cleavage (Arg153–Arg154) [15]. Whether this potential cleavage site is of biological relevance is unclear.

Thrombopoietin (TPO) structure. Amino acids 1–153 define the “EPO-like” domain which binds the TPO receptor. Amino acids 154–332 define the “carbohydrate-rich” domain that provides stability. Sites of potential thrombin cleavage are indicated. Red bars indicate known disulphide linkages (Courtesy of Dr. T. Kato, Pharmaceutical Research Laboratory, Kirin Brewing Company, Takasaki, Gunma, Japan)
The amino acid structure of the EPO-like domain is highly conserved and the first 153 amino acids of TPO are 23 % identical with human EPO and probably 50 % similar if conservative amino acid substitutions are taken into consideration [16]. Furthermore, the EPO domain of TPO contains four alpha-helical regions which are structurally similar to those in EPO. However, EPO does not bind the TPO receptor and, conversely, TPO does not bind the EPO receptor.
The role of the carbohydrate-rich domain of thrombopoietin remains less clear. None of this region is important for receptor binding and this region is less conserved among different species. Murine and human thrombopoietin are only 62 % identical in the carbohydrate-rich domain while they are 84 % identical in the EPO-like domain. While all of the receptor binding activity reside in the EPO domain, the carbohydrate-rich domain seems to be important for stabilizing the molecule in the circulation; indeed the truncated first 153 amino acids of TPO has a markedly decreased circulatory half-life compared to the 20- to 40-h half-life of the mature protein [17]. (Paradoxically the truncated molecule has a specific activity in vitro 20-fold higher than the whole molecule [18, 19].) Presumably, the glycosylated domain of TPO confers stability and prolongs its circulatory half-life just like the way carbohydrate sequences regulate the stability of EPO [19, 20].
Recent analysis of the crystal structure of the receptor-binding domain (residues 1–163) has determined that TPO has an antiparallel four-helix bundle fold and can interact with a soluble thrombopoietin receptor construct with a 1:2 stoichiometry with one high-affinity (3.3 × 109 M−1) and one low-affinity (1.1 × 109 M−1) binding site [21]. This leads to the functional properties which are discussed next.
Thrombopoietin function
The mechanism by which TPO binds to the TPO receptor and initiates signal transduction has been well outlined (Fig. 2). Like the EPO receptor [22, 23], the TPO receptor (c-mpl) probably exists as a preformed but inactive dimer. Each TPO receptor monomer contains two CRH (cytokine receptor homology) domains. In the absence of the distal CRH, the TPO receptor is active suggesting that the distal CRH domain inhibits activation of the TPO receptor until relieved by TPO binding. TPO binds only to the distal CRH of the TPO receptor and not to the proximal CRH and thereby activates the receptor.

Activation of the TPO receptor by TPO or TPO receptor agonists. TPO and romiplostim bind to the distal cytokine receptor homology domain (CRH-2) of the preformed, inactive TPO receptor dimer. Eltrombopag binds to the transmembrane region of the TPO receptor. Multiple signal transduction pathways are thereby activated
Binding of TPO to its receptor initiates a wide variety of signal transduction pathways. The best known of these are the JAK and STAT pathways, which become phosphorylated and promote cell growth [24]. In addition, MAP kinase pathways are activated, which potentiate maturation. Finally, less well studied but equally important, are the anti-apoptotic pathways that are activated [25–27]. As discussed next, TPO binding results in mitosis, endomitosis, maturation, and a wide variety of anti-apoptotic effects in megakaryocyte precursors and in megakaryocytes [6, 28]. Indeed, the removal of TPO from the circulation is also regulated by this receptor binding, most likely on platelets [29, 30].
Thrombopoietin physiology
The thrombopoietin receptor is present in a wide variety of hematopoietic tissues ranging from stem cells to megakaryocyte colony forming cells (Meg-CFC), myeloid and erythroid progenitors, to early and late megakaryocytes, as well as mature platelets. TPO receptors are not found on normal or malignant non-hematopoietic tissues [31], except possibly cardiac myocytes [26] and brain neurons, astrocytes and microglial cells [32].
Each hematopoietic tissue has its own unique response to TPO receptor binding.
-
TPO is necessary for the viability of stem cells. Children and animals born without TPO or the TPO receptor are thrombocytopenic at birth and develop pancytopenia over time [33]. Indeed, patients who develop a neutralizing antibody to thrombopoietin eventually develop pancytopenia [10, 34, 35].
-
TPO stimulates mitosis of Meg-CFC [35].
-
TPO stimulates megakaryocyte endomitosis and increases the average ploidy of megakaryocytes. Of all the effects of TPO, this requires the lowest TPO concentration [6].
-
TPO increases megakaryocyte maturation but seems to have little role in the release of platelets from megakaryocytes [36].
-
TPO is the prime regulator of platelet production. In animals in which both copies of the TPO gene have been eliminated, platelet counts are approximately 10 % of normal but the animals are otherwise healthy [36]. Knockout of only one of the two TPO genes produces animals with half-normal platelet count, confirming a TPO dosage effect. Animals in which both copies of the TPO receptor gene are eliminated are thrombocytopenic; however, animals with loss of one TPO receptor gene have a normal platelet count [37–39].
-
TPO affects early progenitor cells of other lineages. Animals that have lost both copies of the TPO gene are thrombocytopenic and have 5–10 % of the normal number of Meg-CFC; but erythroid and multilineage colony forming cells are also markedly decreased without affecting the red blood cell or white blood cell count [37]. This suggests that in the normal physiology TPO is responsible for the early maturation of all lineages but only for the specific amplification of megakaryocytes.
-
TPO binds with high affinity to platelet TPO receptors. There are 56 ± 17 TPO receptors per platelet with a K D of 163 ± 31 pM and with these the platelets bind, internalize and degrade TPO [29, 30]. This seems to be the primary means by which TPO is regulated in normal physiology [6].
In normal physiology TPO is made at a constant rate in the liver, released into the circulation without any storage form, and then rapidly cleared from the circulation by platelets and possibly also megakaryocytes, leaving a basal level of thrombopoietin in the circulation [6, 40]. There is no cytokine or clinical scenario that increases TPO mRNA or TPO production [41]. After liver resection or liver disease, TPO levels and platelet counts decrease [42, 43]. In situations of reduced platelet production such as after chemotherapy, clearance by platelets and megakaryocytes is decreased, TPO levels increase approximately 10- to 20-fold over baseline, in an apparent effort to increase megakaryocyte growth and platelet production [44]. In contrast, in ITP TPO levels are normal despite the low platelet count, probably because platelet production and the flux of platelets through the circulation are normal or slightly elevated and despite their short half-life total TPO clearance is normal [45, 46]. Furthermore, in patients with essential thrombocythemia (ET) TPO levels are normal because despite the markedly elevated platelet count, ET platelets have a markedly reduced number of TPO receptors and net TPO clearance is normal [47]. Indeed, it has been speculated that the elevated platelet count in this disorder is actually due to this altered TPO clearance mechanism [47].
This crude but effective feedback loop is in contrast to that seen with EPO which has a well-defined mechanism in which the kidney detects hypoxia and alters the transcription of the EPO gene. There does not seem to be any significant increase in TPO gene transcription in the liver in thrombocytopenic animals [40]. This regulation of platelet production by TPO is similar to that by which neutrophil and monocyte production is regulated by G-CSF and M-CSF in normal physiology [48].
Lessons learned from clinical studies with recombinant thrombopoietin molecules
After the initial discovery of thrombopoietin in 1994, two recombinant proteins, rhTPO and PEG-rhMGDF, rapidly entered clinical development. rhTPO was a glycosylated recombinant protein expressed in CHO cells that had an amino acid sequence identical to that of endogenous thrombopoietin although with a slightly different glycosylation pattern. In contrast, PEG-rhMGDF was a truncated non-glycosylated protein containing all of the first 153 amino acids of TPO, the receptor-binding domain, coupled to polyethylene glycol to improve stability [7].
Both of these recombinant proteins had a circulatory half-life of 30–40 h and markedly increased platelet counts in a dose-dependent fashion in healthy volunteers or in cancer chemotherapy patients prior to chemotherapy [8, 49]. These two proteins defined the platelet count response that can be expected after administration of recombinant TPO as well as to the TPO receptor agonists [50]. That is, after administering a single dose of TPO to a healthy animal or human, megakaryocyte ploidy becomes maximal on Day 3 and megakaryocyte numbers increase on Day 4 [51]. There is no increase in the platelet count until Day 5; the platelet count continues to rise until Day 10–14 after which the platelet count gradually drifts back to baseline by Day 28 [50].
In clinical studies, rhTPO was given by an intravenous injection and PEG-rhMGDF as a subcutaneous injection. Both molecules were studied in a wide variety of clinical situations. In lung cancer [8] or gynecological cancer [52] chemotherapy trials, administration of a recombinant TPO raised the nadir platelet count, decreased the duration of thrombocytopenia, and in some situations reduced the need for platelet transfusions. However, in myeloablative chemotherapy regimens (e.g., stem cell transplantation, leukemia induction chemotherapy) administration of either recombinant protein before, during and/or after the chemotherapy failed to show any improvement in platelet independence over 20 × 109, duration of thrombocytopenia, or need for platelet transfusions [53, 54]. Both molecules produced a profound rebound thrombocytosis upon platelet recovery which was clinically irrelevant.
In a few patients with myelodysplastic syndrome, administration of recombinant TPO was associated with an increase in platelet count and in some a multilineage response was seen [55]. Three of four ITP patients given PEG-rhMGDF for 7 days had their platelet count rise to normal [9]. In patients being prepared for autologous stem cell transplantation, priming of the patient with rhTPO plus G-CSF increased the yield of CD34 cells collected at apheresis over those treated with just G-CSF. There was a statistically significant improvement in engraftment with rhTPO mobilization but the magnitude of the effect was clinically not that meaningful [53].
Finally, PEG-rhMGDF was given to a large number of healthy platelet apheresis donors. Treatment with a single dose produced a dose-dependent increase in platelet count and also in the apheresis yield. Yields rose from 3.3 × 1011 platelets per donation in the placebo group to a high of 10.6 × 1011 in those receiving the highest dose of PEG-rhMGDF [56]. These platelets were functional and, when transfused to a thrombocytopenic patients undergoing myeloablative therapy, produced a dose-dependent rise in platelet count [57–59]. Similar results were seen in studies with rhTPO [60].
In preparation for larger studies in apheresis patients, a large number of paid healthy human volunteers were administered PEG-rhMGDF. These subjects had the expected rise in platelet count, but 13 of 535 developed paradoxical thrombocytopenia due to the production of an autoantibody that neutralized PEG-rhMGDF and also cross-reacted with and neutralized the endogenous TPO [10]. Fortunately, all of these healthy subjects recovered from their thrombocytopenia upon drug cessation; a few required a brief course of immunosuppressive therapy. None have demonstrated long-term complications from this exposure. Thrombocytopenia was never reported with rhTPO but by 2001 development of both recombinant proteins had stopped [61].
TPO receptor agonists
The overall encouraging results of clinical work with recombinant thrombopoietins spurred development of new thrombopoietic agents that were not immunogenic and might have improved properties, such as oral administration (Table 1). Out of these efforts have come two TPO receptor agonists, romiplostim and eltrombopag [11, 12, 62–64].
Romiplostim
In 1994, Cwirla used a random peptide library to screen for peptides that bound to an immobilized 484 amino acid extracellular domain of the TPO receptor [65, 66]. A core consensus peptide was identified that had no homology with endogenous TPO and this was further modified to enhance its binding to the TPO receptor. The 14 amino acid peptide (IEGPTLRQWLAARA) thus created had a high-binding affinity for the TPO receptor and when dimerized by a β-alanine bridge created a dimeric peptide that was ~4,000-fold more active, comparable to that of recombinant TPO in stimulating the growth of a TPO-dependent cell line.
The key attributes of this peptide construct were its unique sequence and its dimeric structure. The former attribute was expected to reduce its immunogenicity since it had no sequence homology with endogenous TPO. The latter attribute would replicate the two different TPO receptor binding sites on TPO and allow it to activate the TPO receptor.
Unfortunately, such peptide constructs have a very short circulatory half-life which markedly limits their in vivo activity [65]. Efforts to prolong the half-life of this peptide were undertaken, the most efficient of which was to insert a pair of these dimeric peptides into the carboxy terminus of each arm of an IgG1 heavy chain, creating what is called a “peptibody” [66]. Crucial aspects of design of this peptibody were to optimize the lengths of the polyglycine spacers that separate the dimeric peptides as well as the lengths of the polyglycine spacers that then join them to the IgG heavy chain.
This is the origin of romiplostim (AMP-2, AMG-531, Nplate) (Fig. 3). The 14-amino acid peptide has no sequence homology with native TPO hoping to minimize the autoantibody risk of prior recombinant products. Romiplostim binds to the TPO receptor just like endogenous TPO but with one-fourth the affinity (Li et al. 2001, unpublished results) and competes with endogenous TPO for binding [67]. Like other IgG molecules, romiplostim is internalized by the FcRn receptor on endothelial cells, protected from degradation and recycled back into the circulation, producing a circulatory half-life of some 120–140 h [68].

In studies in vitro and in vivo, romiplostim has the same mechanism of action as does recombinant TPO. Romiplostim stimulates the growth of Meg-CFC, increases the number, size, and ploidy of megakaryocytes [67], and can enhance the production of platelets in an in vitro culture system [69].
After a single subcutaneous romiplostim dose is administered to healthy volunteers, there is the typical platelet count response seen with all TPO molecules: there is no increase until Day 5, after which there is a dose-dependent rise in platelet count that peaks on Days 12–14, followed by a decline to baseline by Day 28[68]. There is a linear dose–response relationship between the dose of romiplostim and the increase in the platelet count, with a maximum platelet count eightfold baseline occurring at a dose of 10 μg/kg.
The clinical pharmacology of romiplostim has been well studied and is summarized in Table 2. Romiplostim is recycled by the FcRn receptor on endothelial cells and terminally cleared by the reticuloendothelial system. There is no known effect of renal or hepatic dysfunction on its half-life or function. Its half-life is 120–140 h independent of whether given by intravenous or subcutaneous routes; it is only formulated for subcutaneous administration. Given the fact that it can cross the placenta by the FcRn receptor, its use in pregnancy is not suggested. Romiplostim is formulated in 250- and 500-μg vials each containing 375 or 625 μg of romiplostim, respectively. When reconstituted, it is administered subcutaneously at a dose of 1–10 μg/kg weekly for the treatment of ITP [70].
The current romiplostim prescribing information for treating ITP recommends a starting dose of 1 μg/kg to be increased by 1 μg/kg weekly to a maximum of 10 μg/kg weekly in order to attain a target platelet count 50–250 × 109/L. It is also recommended that the weekly dose be held if the platelet count exceeds 400 × 109/L. It is the opinion and experience of this author that these two recommendations are inappropriate and may adversely affect patient safety. Since the effective average dose of romiplostim in most studies was 4–5 μg/kg [45, 71–73], this author recommends starting at 3 μg/kg in most settings and increasing by 2 μg/kg weekly until the desired platelet count is attained. In more urgent settings even higher starting doses, including the maximal dose of 10 μg/kg, may be used to determine rapidly if this agent will be effective in the patient. In addition, holding romiplostim for platelet counts over 400 × 109/L is not recommended; it may result in a rapid and potentially dangerous decrease in platelet count within a week. This author has commonly seen platelet counts drop from 1,000 × 109/L to 3 × 109/L in under a week when weekly dosing was held. Instead of holding the dose, this author recommends a dose decrease of 25–50 % if the platelet count rises to over 400 × 109/L. When the platelet count rise is associated with corticosteroid administration, no dose change is recommended; corticosteroids synergize strongly all TPO receptor agonists. For extreme elevations of platelets over 800 × 109/L, this author routinely administers aspirin 81 mg daily.
Eltrombopag
High throughput screening for small molecules that stimulate the TPO receptor identified a number of non-peptide chemical agents that bind and activate this receptor. A number of hydrazinonaphthalene derivatives [74–77] were initially identified and these lead compounds were modified to enhance their pharmacologic and biologic properties [78, 79]. One molecule which evolved from this process was eltrombopag (SB-497115, Promacta, Revolade). This is a 442 Da molecule that is administered orally once daily (Fig. 4).

Eltrombopag structure. Eltrombopag [3′-{N’-[1-(3,4-dimethyl-phenyl)-3-methyl-5-oxo-1,5-dihydropyrazol-4-ylidene]hydrazino}-2′-hydroxybiphenyl-3-carboxylic acid] has an acidic (COOH) group at one end, lipophilic (CH3) groups at the other end, and a metal chelate group in the center that creates a potent, orally available TPO non-peptide agonist. (Courtesy of Dr. Richard J. Frankovitch and Julian Jenkins, GlaxoSmithKline, Collegeville, PA.)
A major difficulty in developing eltrombopag was the fact that it does not have any biological activity in species other than humans and chimpanzees [80]. Assessment of the reason for this species specificity has identified an interesting mechanism of action for eltrombopag. At residue 499 in the transmembrane region of the TPO receptor of humans and chimpanzees there is a histidine, whereas all other species have a leucine. If one converts the histidine to a leucine in the human receptor, there is no response to eltrombopag. Conversely, if one changes the murine receptor leucine to a histidine, the cell now responds to eltrombopag. The threonine in position 496 is also unique in humans and chimpanzees and may also be important. It therefore appears that eltrombopag binds to the transmembrane region of the TPO receptor and activates it by a mechanism different from that of recombinant TPO or romiplostim [12].
There are a number of important implications of the finding that eltrombopag activates the TPO receptor different from recombinant TPO or romiplostim (Fig. 2).
-
Eltrombopag does not compete with thrombopoietin for binding to the distal cytokine homology region of the TPO receptor; both molecules can potentially bind the same receptor, but at different locations. In both in vitro and in vivo settings, the effects of eltrombopag are additive to the effects of thrombopoietin (Fig. 5) [81]. This may be clinically relevant in that it might predict activity of eltrombopag in situations (e.g., myeloablative chemotherapy) where endogenous TPO levels are already high and in which adding more recombinant TPO or romiplostim might have not have additional benefit [82].
Fig. 5 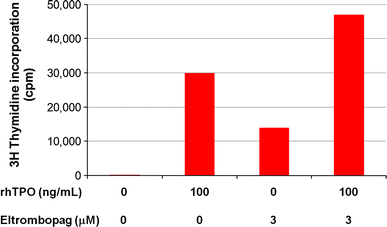
Eltrombopag effect is additive to that of rhTPO. Growth of a TPO-dependent cell line is measured by the incorporation of 3H-thymidine into DNA. At maximal doses the stimulatory effect of eltrombopag is less than that of rhTPO; however, together there is an additive effect. Redrawn from data in Erickson-Miller et al. [81]
-
Eltrombopag activates signal transduction pathways differently than TPO or romiplostim [83]. Eltrombopag is a much weaker stimulator of JAK and STAT phosphorylation. Eltrombopag does not activate the AKT pathway at all, unlike recombinant TPO or romiplostim.
-
Eltrombopag does not potentiate platelet activation by platelet agonists such as ADP in standard aggregometry experiments, probably due to the altered signal transduction pathways mentioned above [83]. Although probably not clinically relevant, TPO and romiplostim both reduce by 50 % the concentration of agonist necessary to activate platelets [84].

When the maximal dose of 75 mg is administered daily to healthy humans for 10 days, eltrombopag produces a maximal dose-dependent rise in platelet counts of 150 × 109/L over baseline [85]. This is one-eighth of the maximal effect produced by romiplostim in healthy subjects, but to date this lower potency has not translated into any difference in clinical response.
The clinical pharmacology of eltrombopag has been well studied (Table 3). Data from 568 subjects that showed that oral administration results in a peak concentration in 2–6 h and excretion half-life of 21–32 h [86]. Sixty percent is hepatically metabolized and 31 % is renally excreted. Plasma levels, therefore, increase in proportion to the reduction in hepatic function. The effects of renal insufficiency have not been studied, but this investigator has not noticed any difference in effect in ITP patients who are on dialysis.
A number of pharmacologic interactions need to be taken into consideration when using eltrombopag. The plasma concentration of eltrombopag is decreased by ~70 % by the presence of divalent cations such as calcium. Eltrombopag inhibits the organic anion transporting peptide OATP1B1; this results in the need to decrease the dose of other concomitant medications metabolized by that pathway, such as statins. Finally, the eltrombopag AUC is 70–80 % higher in patients of East Asian ancestry mandating a decreased starting dose for such patients.
Eltrombopag is formulated as eltrombopag olamine in pill sizes of 12.5, 25, 50, 75 and 100 mg to be taken orally once a day. For ITP patients, the starting dose is 50 mg (25 mg if East Asian or if significant hepatic insufficiency). Eltrombopag is taken on an empty stomach 1 h before or 2 h after a meal with no divalent cations being coadministered within 4 h. Finally, since approximately 13 % of patients developed mild liver function abnormalities, occasional liver function tests are warranted [87].
Clinical development of TPO receptor agonists
Both eltrombopag and romiplostim are currently approved by many regulatory agencies worldwide for the treatment of thrombocytopenia in ITP patients who failed splenectomy or those unable to undergo that procedure. This is based upon a large number of clinical trials that have shown that the administration of either TPO receptor agonist increases the platelet count (Fig. 6), decreases the need for rescue medications, decreases the need for other ITP therapies such as corticosteroids, may avoid the need for splenectomy, improves the quality of life, and markedly decreases the rate of bleeding [45, 71, 87–89].

Romiplostim increases the platelet count in ITP patients. Platelet count (upper panel) and romiplostim dose (lower panel) in an ITP patient treated for over 5 years. Note that romiplostim was administered at a constant weekly dose of 2 μg/kg but that the platelet counts were measured every 1–3 months. The significant month-to-month fluctuations are typical of ITP patients on many forms of chronic treatment including corticosteroids, azathioprine, danazol, and TPO receptor agonists
Pediatric ITP patients respond to TPO receptor agonists as well as, if not better than, adult ITP patients [90]. Given the fact that many pediatric ITP patients require therapy only for a short period of time, this might become a standard of care for this patient group since the risk profile of these agents over this short timeframe is well understood.
Eltrombopag has also recently been approved in the United States for the treatment of thrombocytopenia due to hepatitis C infection in patients who are undergoing anti-viral treatment. This is based upon clinical trials showing that eltrombopag increased the platelet count of such patients, reduced the subsequent extent of thrombocytopenia on anti-viral treatment, and allowed most patients to continue antiviral therapy that would otherwise not have been possible [91].
Treatment with TPO receptor agonists has also been studied in a wide range of other thrombocytopenic conditions. As discussed elsewhere in this compendium, eltrombopag increased not only the platelet count but also sometimes other lineages when given to aplastic anemia patients [92]. Doses were often higher than those used to treat ITP. Considering that endogenous TPO levels in these patients were already elevated 10–20 fold above normal, it is unclear how eltrombopag is working. Since eltrombopag enhances the effect of TPO in vitro [81], it is possible that it is doing so in vivo; or there is a totally different mechanism of action which has not yet been uncovered.
Eltrombopag increased the platelet count in 11 of 12 patients with myosin heavy chain-9 (MYH9)-related thrombocytopenia. Although this condition is not one that commonly causes significant hemorrhage, the bleeding tendency disappeared in 8 of 10 subjects with bleeding at baseline [93, 94].
A major focus of the first generation recombinant thrombopoietins was chemotherapy-induced thrombocytopenia. This still remains an area of interest for the TPO receptor agonists, but no major study has yet been reported. A number of small studies have shown modest improvement in platelet counts with the administration of either romiplostim or eltrombopag [95–98]. None of these studies is large enough or sufficiently well constructed to allow a clear message. Since both TPO receptor agonists function like the earlier recombinant thrombopoietins, this author believes that adequate studies will eventually be done to demonstrate that TPO receptor agonists will increase the nadir platelet count, decrease the need for platelet transfusions and allow for chemotherapy to be given at full dose and on schedule.
Adverse events, observed and potential
Table 4 lists the observed and potential adverse consequences of eltrombopag and romiplostim. Almost all these data come from ITP patients studied in two recent long-term safety studies [71, 87].
Observed adverse events
Rebound worsening of thrombocytopenia In ITP patients, the platelet count usually returns to its prior baseline upon discontinuation of TPO receptor agonists [45, 89]. However, in approximately 10 % of such patients the platelet count drops below its prior baseline for 1–3 weeks before going back up to baseline [45]. This rebound worsening of thrombocytopenia is of concern and despite statements in the prescribing information for both drugs that drug should be held when the platelet count rises over 400 × 109/L, this author cautions against this recognizing that the platelet count may abruptly decline to below 10 × 109/L within 5–7 days and expose some patients to a major bleeding risk. Rather, this author suggests that the dose be reduced by 25–50 % in this setting. Even though there is no evidence that an elevated platelet count is harmful in this setting, for extreme platelet count elevations to over 800 × 109/L occasionally seen, this author will often administer aspirin 81 mg daily. This concern of rebound thrombocytopenia applies only to ITP patients.
Increased bone marrow reticulin In acute myeloid leukemia patients treated with rhTPO, eight patients developed increased reticulin but this disappeared in all within a month of drug discontinuation [99]. In animals exposed to romiplostim, there was a dose-dependent rise in reticulin which disappeared after 1 month off romiplostim [100]. In clinical studies of romiplostim [71, 100], only 38 of 292 patients had a bone marrow examination, 11 of which had increased reticulin. When compared to the 27 patients without increased reticulin, these 11 had no chromosomal abnormalities, less response to therapy, and a higher rate of splenectomy. In two patients who had subsequent bone marrow biopsies off drug, the reticulin disappeared. In an ongoing 3-year prospective bone marrow study of ITP patients being treated with romiplostim, at the 1-year time point, there was no increase in collagen (trichrome) staining and only 2/38 had increased reticulin (but still within the normal range) [101].
In a study of 110 ITP patients treated with eltrombopag for a median 18 months and who had 156 evaluable bone marrow examinations, 152/156 (97.4 %) lacked any increased reticulin; only 4 (2.6 %) biopsies from 3 patients showed increased reticulin and 3 biopsies (from the same three patients) also showed increased collagen [102]. Of the 3 patients, 2 discontinued eltrombopag and 1 had a marrow examination done 7 months later that was normal. The third patient continued on therapy despite having increased collagen and reticulin fibrosis. A prospective, 2-year bone marrow study is ongoing to assess bone marrow fibrosis [103].
Thrombocytosis Both eltrombopag and romiplostim can promote thrombocytosis with platelet counts sometimes rising to over 1,000 × 109/L. In other clinical settings, there is no evidence that thrombocytosis is associated with thrombotic risk, even in patients with myeloproliferative diseases. As mentioned above, the prescribing information suggests holding either drug if platelet counts are over 400 × 109/L, but this is based on no clinical data suggesting a detrimental outcome, including thrombosis (vide infra).
Stimulation of growth of leukemic blasts A new concern for TPO receptor agonists has come from a recent study of romiplostim in myelodysplastic syndrome (MDS). Patients with IPSS low/intermediate-1 risk MDS, with platelet counts less than 20 × 109/L or 50 × 109/L, were given romiplostim 750 weekly (167 patients) versus placebo (83 patients) [104]. A platelet response was seen in 37 % of patients given romiplostim and less than 4 % of patients on placebo. Bleeding was modestly reduced from 27 % in placebo to 19 % in romiplostim patients while transfusion events decreased from 1014 to 749 per 100 patient-weeks. But this study was stopped early because 6 % (10/167) of patients receiving romiplostim versus 4.8 % (4/83) of placebo patients showed a possible progression to AML. Unfortunately, the conversion to leukemia was not documented by marrow examination. Of equal concern was the effect on the percentage of patients with circulating blasts over 10 %; this occurred in 15 % of romiplostim patients versus 3.6 % of placebo patients. However, upon discontinuing the romiplostim, 20 of the 28 showed no progression of AML. These studies do provide pause for the use of either TPO receptor agonist in patients with MDS. What is of some interest, however, is the fact that the first generation TPO agents were given to patients with acute leukemia with no apparent affect upon their remission or relapse rates [7].
Reduction in threshold for platelet activation Romiplostim does reduce by 50 % the threshold for platelet activation by weak agonists such as ADP in standard platelet aggregometry studies [50]. Eltrombopag lacks this effect [83]. Whether this is important clinically or not is unclear, but to date there has been no increased risk of thrombosis with this agent. rhTPO and PEG-rhMGDF produced identical effects but in several animal models of thrombosis, no increased thrombotic effect was noted [50].
Potential adverse events
Thrombosis In placebo-controlled ITP trials, thrombosis has not been an issue with either TPO receptor agonist. The thrombosis rate with romiplostim in ITP patients is approximately 0.09 events per 100 patient-weeks which is identical to the 0.10 events per 100 patient-weeks seen with placebo [45]. With long-term romiplostim treatment, the thrombosis rate is 0.08 events per 100 patient-weeks [71]. In those with thrombosis, there was no association with the platelet count; thrombosis occurred at platelet counts as low as 6 × 109/L and as high as 1,359 × 109/L. Identical results have come from recent long-term analyses of eltrombopag which have shown no relationship of thrombosis with platelet count [87].
This data should be evaluated in the context of the recent understanding that ITP is a prothrombotic disorder with a rate of thrombosis that is approximately 1.41 times that of a comparable non-ITP patient population matched for age and gender [105]. Cumulative rates as high as 6.9 % over a 15-month period have been reported in adults [106] and a recent Danish registry showed a relative risk of 2.65 over a control population without ITP [107].
The only possible exception comes from data in chronic liver disease patients receiving eltrombopag to increase the platelet count in preparation for surgical procedures [108]. Although baseline vascular assessments were unfortunately not done, 6/145 patients receiving eltrombopag and 1/147 patients receiving placebo developed symptomatic portal vein thrombosis. However, some patients had other risk factors for thrombosis, such as the presence of hepatocellular carcinoma.
Stimulation of growth of solid tumors The recent concern that EPO may stimulate tumor cell growth is probably not a risk for TPO receptor agonists. The TPO receptor is not found on non-hematological solid tumors [31]. In a large number of solid tumor chemotherapy studies with rhTPO and PEG-rhMGDF there was no enhancement of tumor growth [7].
Antibody formation Antibody formation was a major concern with the recombinant TPO, PEG-rhMGDF [10, 34]. To date no antibodies have been formed against eltrombopag, and only two patients have developed non-neutralizing antibody against romiplostim [71]. Indeed, these two antibodies were identified within the first 100 patients treated with romiplostim and the antibody disappeared at later evaluation with no apparent effect on the platelet count. Subsequently no antibodies have been identified.
Stem cell depletion This theoretical concern has also been raised after long-term administration of erythropoietin and G-CSF, but has not been reported. Indeed, families with overexpression of the TPO gene have not shown any evidence for stem cell depletion [109, 110].
Interactions with other cytokines or hematopoietic growth factors This has not been observed.
References
Kelemen E, Cserhati I, Tanos B. Demonstration and some properties of human thrombopoietin in thrombocythaemic sera. Acta Haematol. 1958;20:350–5.
de Sauvage FJ, Hass PE, Spencer SD, et al. Stimulation of megakaryocytopoiesis and thrombopoiesis by the c-Mpl ligand. Nature. 1994;369:533–8.
Lok S, Kaushansky K, Holly RD, et al. Cloning and expression of murine thrombopoietin cDNA and stimulation of platelet production in vivo. Nature. 1994;369:565–8.
Bartley TD, Bogenberger J, Hunt P, et al. Identification and cloning of a megakaryocyte growth and development factor that is a ligand for the cytokine receptor Mpl. Cell. 1994;77:1117–24.
Kato T, Ogami K, Shimada Y, et al. Purification and characterization of thrombopoietin. J Biochem (Tokyo). 1995;118:229–36.
Kuter DJ, Beeler DL, Rosenberg RD. The purification of megapoietin: a physiological regulator of megakaryocyte growth and platelet production. Proc Natl Acad Sci (USA). 1994;91:11104–8.
Kuter DJ, Begley CG. Recombinant human thrombopoietin: basic biology and evaluation of clinical studies. Blood. 2002;100:3457–69.
Fanucchi M, Glaspy J, Crawford J, et al. Effects of polyethylene glycol-conjugated recombinant human megakaryocyte growth and development factor on platelet counts after chemotherapy for lung cancer. N Engl J Med. 1997;336:404–9.
Nomura S, Dan K, Hotta T, Fujimura K, Ikeda Y. Effects of pegylated recombinant human megakaryocyte growth and development factor in patients with idiopathic thrombocytopenic purpura. Blood. 2002;100:728–30.
Li J, Yang C, Xia Y, et al. Thrombocytopenia caused by the development of antibodies to thrombopoietin. Blood. 2001;98:3241–8.
Kuter DJ. New thrombopoietic growth factors. Blood. 2007;109:4607–16.
Kuter DJ. Thrombopoietin and thrombopoietin mimetics in the treatment of thrombocytopenia. Annu Rev Med. 2009;60:193–206.
Foster DC, Sprecher CA, Grant FJ, et al. Human thrombopoietin: gene structure, cDNA sequence, expression, and chromosomal localization. Proc Natl Acad Sci USA. 1994;91:13023–7.
Hoffman RC, Andersen H, Walker K, et al. Peptide, disulfide, and glycosylation mapping of recombinant human thrombopoietin from ser1 to Arg246. Biochemistry (Mosc). 1996;35:14849–61.
Kato T, Oda A, Inagaki Y, et al. Thrombin cleaves recombinant human thrombopoietin: one of the proteolytic events that generates truncated forms of thrombopoietin. Proc Natl Acad Sci (USA). 1997;94:4669–74.
Gurney AL, Kuang WJ, Xie MH, Malloy BE, Eaton DL, de Sauvage FJ. Genomic structure, chromosomal localization, and conserved alternative splice forms of thrombopoietin. Blood. 1995;85:981–8.
Hokom MM, Lacey D, Kinstler OB, et al. Pegylated megakaryocyte growth and development factor abrogates the lethal thrombocytopenia associated with carboplatin and irradiation in mice. Blood. 1995;86:4486–92.
Foster D, Hunt P. The biological significance of truncated and full-length forms of Mpl ligand. In: Kuter DJ, Hunt P, Sheridan W, Zucker-Franklin D, editors. Thrombopoiesis and thrombopoietins: molecular, cellular, preclinical, and clinical biology. Totowa: Humana Press; 1997. p. 203–14.
Foster D, Lok S. Biological roles for the second domain of thrombopoietin. Stem Cells. 1996;14(Suppl 1):102–7.
Spivack JL, Hogans BB. The in vivo metabolism of recombinant human erythropoietin in the rat. Blood. 1989;73:90–9.
Feese MD, Tamada T, Kato Y, et al. Structure of the receptor-binding domain of human thrombopoietin determined by complexation with a neutralizing antibody fragment. Proc Natl Acad Sci USA. 2004;101:1816–21.
Remy I, Wilson IA, Michnick SW. Erythropoietin receptor activation by a ligand-induced conformation change. Science. 1999;283:990–3.
Livnah O, Stura EA, Middleton SA, Johnson DL, Jolliffe LK, Wilson IA. Crystallographic evidence for preformed dimers of erythropoietin receptor before ligand activation. Science. 1999;283:987–90.
Drachman JG, Griffin JD, Kaushansky K. The c-Mpl ligand (thrombopoietin) stimulates tyrosine phosphorylation of Jak2, Shc, and c-Mpl. J Biol Chem. 1995;270:4979–82.
Hoffmann O, Rung O, Im AR, et al. Thrombopoietin contributes to neuronal damage in experimental bacterial meningitis. Infect Immun. 2011;79:928–36.
Baker JE, Su J, Hsu A, et al. Human thrombopoietin reduces myocardial infarct size, apoptosis, and stunning following ischaemia/reperfusion in rats. Cardiovasc Res. 2008;77:44–53.
Borge OJ, Ramsfjell V, Veiby OP, Murphy MJ Jr, Lok S, Jacobsen SE. Thrombopoietin, but not erythropoietin promotes viability and inhibits apoptosis of multipotent murine hematopoietic progenitor cells in vitro. Blood. 1996;88:2859–70.
Zauli G, Vitale M, Falcieri E, et al. In vitro senescence and apoptotic cell death of human megakaryocytes. Blood. 1997;90:2234–43.
Li J, Xia Y, Kuter D. Interaction of thrombopoietin with the platelet c-mpl receptor in plasma: binding, internalization, stability and pharmacodynamics. Br J Haematol. 1999;106:345–56.
Broudy VC, Lin NL, Sabath DF, Papayannopoulou T, Kaushansky K. Human platelets display high-affinity receptors for thrombopoietin. Blood. 1997;89:1896–904.
Columbyova L, Loda M, Scadden DT. Thrombopoietin receptor expression in human cancer cell lines and primary tissues. Cancer Res. 1995;55:3509–12.
Zhang J, Freyer D, Rung O, Im AR, Hoffmann O, Dame C. Inflammation stimulates thrombopoietin (Tpo) expression in rat brain-derived microvascular endothelial cells, but suppresses Tpo in astrocytes and microglia. J Interferon Cytokine Res. 2010;30:465–9.
Ballmaier M, Germeshausen M, Schulze H, et al. c-mpl mutations are the cause of congenital amegakaryocytic thrombocytopenia. Blood. 2001;97:139–46.
Basser RL, O’Flaherty E, Green M, et al. Development of pancytopenia with neutralizing antibodies to thrombopoietin after multicycle chemotherapy supported by megakaryocyte growth and development factor. Blood. 2002;99:2599–602.
Kaushansky K, Broudy VC, Grossmann A, et al. Thrombopoietin expands erythroid progenitors, increases red cell production, and enhances erythroid recovery after myelosuppressive therapy. J Clin Invest. 1995;96:1683–7.
de Sauvage FJ, Carver-Moore K, Luoh SM, et al. Physiological regulation of early and late stages of megakaryocytopoiesis by thrombopoietin. J Exp Med. 1996;183:651–6.
Carver-Moore K, Broxmeyer HE, Luoh SM, et al. Low levels of erythroid and myeloid progenitors in thrombopoietin- and c-mpl-deficient mice. Blood. 1996;88:803–8.
de Sauvage FJ, Villeval JL, Shivdasani RA. Regulation of megakaryocytopoiesis and platelet production: lessons from animal models. J Lab Clin Med. 1998;131:496–501.
Gurney AL, Carver-Moore K, de Sauvage FJ, Moore MW. Thrombocytopenia in c-mpl-deficient mice. Science. 1994;265:1445–7.
Yang C, Li J, Kuter DJ. The physiological response of thrombopoietin (c-Mpl ligand) to thrombocytopenia in the rat. Br J Haematol. 1999;105:478–85.
Stoffel R, Wiestner A, Skoda RC. Thrombopoietin in thrombocytopenic mice: evidence against regulation at the mRNA level and for a direct regulatory role of platelets. Blood. 1996;87:567–73.
Peck-Radosavljevic M, Wichlas M, Zacherl J, et al. Thrombopoietin induces rapid resolution of thrombocytopenia after orthotopic liver transplantation through increased platelet production. Blood. 2000;95:795–801.
Peck-Radosavljevic M, Zacherl J, Meng YG, et al. Is inadequate thrombopoietin production a major cause of thrombocytopenia in cirrhosis of the liver? J Hepatol. 1997;27:127–31.
Nichol JL. Thrombopoietin levels after chemotherapy and in naturally occurring human diseases. Curr Opin Hematol. 1998;5:203–8.
Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet. 2008;371:395–403.
Kuter DJ. The physiology of platelet production. Stem Cells. 1996;14:88–101.
Li J, Xia Y, Kuter DJ. The platelet thrombopoietin receptor number and function are markedly decreased in patients with essential thrombocythaemia. Br J Haematol. 2000;111:943–53.
Bartocci A, Mastrogiannis DS, Migliorati G, Stockert RJ, Wolkoff AW, Stanley ER. Macrophages specifically regulate the concentration of their own growth factor in the circulation. Proc Natl Acad Sci USA. 1987;84:6179–83.
Vadhan-Raj S, Murray LJ, Bueso-Ramos C, et al. Stimulation of megakaryocyte and platelet production by a single dose of recombinant human thrombopoietin in patients with cancer. Ann Int Med. 1997;126:673–81.
Harker LA, Marzec UM, Hunt P, et al. Dose-response effects of pegylated human megakaryocyte growth and development factor on platelet production and function in nonhuman primates. Blood. 1996;88:511–21.
Arnold JT, Daw NC, Stenberg PE, Jayawardene D, Srivastava DK, Jackson CW. A single injection of pegylated murine megakaryocyte growth and development factor (MGDF) into mice is sufficient to produce a profound stimulation of megakaryocyte frequency, size, and ploidization. Blood. 1997;89:823–33.
Vadhan-Raj S, Verschraegen CF, Bueso-Ramos C, et al. Recombinant human thrombopoietin attenuates carboplatin-induced severe thrombocytopenia and the need for platelet transfusions in patients with gynecologic cancer. Ann Intern Med. 2000;132:364–8.
Somlo G, Sniecinski I, ter Veer A, et al. Recombinant human thrombopoietin in combination with granulocyte colony-stimulating factor enhances mobilization of peripheral blood progenitor cells, increases peripheral blood platelet concentration, and accelerates hematopoietic recovery following high-dose chemotherapy. Blood. 1999;93:2798–806.
Schiffer CA, Miller K, Larson RA, et al. A double-blind, placebo-controlled trial of pegylated recombinant human megakaryocyte growth and development factor as an adjunct to induction and consolidation therapy for patients with acute myeloid leukemia. Blood. 2000;95:2530–5.
Komatsu N, Okamoto T, Yoshida T, et al. Pegylated recombinant human megakaryocyte growth and development factor (PEG-rHuMGDF) increased platelet counts (plt) in patients with aplastic anemia (AA) and myelodysplastic syndrome (MDS). Blood. 2000;96:296a.
Kuter DJ, Goodnough LT, Romo J, et al. Thrombopoietin therapy increases platelet yields in healthy platelet donors. Blood. 2001;98:1339–45.
Goodnough LT, Kuter DJ, McCullough J, et al. Prophylactic platelet transfusions from healthy apheresis platelet donors undergoing treatment with thrombopoietin. Blood. 2001;98:1346–51.
Goodnough LT, DiPersio J, McCullough J, et al. Pegylated recombinant human megakaryocyte growth and development factor (PEG-rHuMGDF) increases platelet (PLT) count (CT) and apheresis yields of normal PLT donors:initial results. Transfusion (Paris). 1997;37:266S.
Kuter DJ. The use of PEG-rhuMGDF in platelet apheresis. Stem Cells. 1998;16:231–42.
Vadhan-Raj S, Kavanagh JJ, Freedman RS, et al. Safety and efficacy of transfusions of autologous cryopreserved platelets derived from recombinant human thrombopoietin to support chemotherapy-associated severe thrombocytopenia: a randomised cross-over study. Lancet. 2002;359:2145–52.
Kuter DJ. Whatever happened to thrombopoietin? Transfusion (Paris). 2002;42:279–83.
Kuter DJ. New drugs for familiar therapeutic targets: thrombopoietin receptor agonists and immune thrombocytopenic purpura. Eur J Haematol Suppl. 2008;69:9–18.
Kuter DJ. New thrombopoietic growth factors. Clin Lymphoma Myeloma. 2009;9(Suppl 3):S347–56.
Kuter DJ. Biology and chemistry of thrombopoietic agents. Semin Hematol. 2010;47:243–8.
Cwirla SE, Balasubramanian P, Duffin DJ, et al. Peptide agonist of the thrombopoietin receptor as potent as the natural cytokine. Science. 1997;276:1696–9.
Molineux G. The development of romiplostim for patients with immune thrombocytopenia. Ann NY Acad Sci. 2011;1222:55–63.
Broudy VC, Lin NL. AMG531 stimulates megakaryopoiesis in vitro by binding to Mpl. Cytokine. 2004;25:52–60.
Wang B, Nichol JL, Sullivan JT. Pharmacodynamics and pharmacokinetics of AMG 531, a novel thrombopoietin receptor ligand. Clin Pharmacol Ther. 2004;76:628–38.
Thon JN, Italiano JE. Platelet formation. Semin Hematol. 2010;47:220–6.
Nplate prescribing information: Amgen, Inc; 2012.
Kuter DJ, Bussel JB, Newland A, et al. Long-term treatment with romiplostim in patients with chronic immune thrombocytopenia: safety and efficacy. Br J Haematol. 2013;161:411–23.
Kuter DJ, Rummel M, Boccia R, et al. Romiplostim or standard of care in patients with immune thrombocytopenia. N Engl J Med. 2010;363:1889–99.
Bussel JB, Kuter DJ, George JN, et al. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med. 2006;355:1672–81.
Duffy KJ, Darcy MG, Delorme E, et al. Hydrazinonaphthalene and azonaphthalene thrombopoietin mimics are nonpeptidyl promoters of megakaryocytopoiesis. J Med Chem. 2001;44:3730–45.
Duffy KJ, Erickson-Miller C. The discovery of eltrombopag, an orally bioavailable TpoR agonist. In: Metcalf D, Dillon SB, editors. Target validation in drug discovery. Burlington: Academic Press; 2007. p. 241–54.
Duffy KJ, Price AT, Delorme E, et al. Identification of a pharmacophore for thrombopoietic activity of small, non-peptidyl molecules. 2. Rational design of naphtho[1,2-d]imidazole thrombopoietin mimics. J Med Chem. 2002;45:3576–8.
Duffy KJ, Shaw AN, Delorme E, et al. Identification of a pharmacophore for thrombopoietic activity of small, non-peptidyl molecules. 1. Discovery and optimization of salicylaldehyde thiosemicarbazone thrombopoietin mimics. J Med Chem. 2002;45:3573–5.
Erickson-Miller C, Delorme E, Giampa L, et al. Biological activity and selectivity for Tpo receptor of the orally bioavailable, small molecule Tpo receptor agonist, SB-497115. Blood. 2004;104:796a.
Erickson-Miller CL, DeLorme E, Tian SS, et al. Discovery and characterization of a selective, nonpeptidyl thrombopoietin receptor agonist. Exp Hematol. 2005;33:85–93.
Erickson-Miller C, Delorme E, Iskander M, et al. Species specificity and receptor domain interaction of a small molecule TPO receptor agonist. Blood. 2004;104:795a.
Erickson-Miller CL, Delorme E, Tian SS, et al. Preclinical activity of eltrombopag (SB-497115), an oral, nonpeptide thrombopoietin receptor agonist. Stem Cells. 2009;27:424–30.
Kuter DJ. What is the potential for thrombopoietic agents in acute leukemia? Best Pract Res Clin Haematol. 2011;24:553–8.
Erhardt JA, Erickson-Miller CL, Aivado M, Abboud M, Pillarisetti K, Toomey JR. Comparative analyses of the small molecule thrombopoietin receptor agonist eltrombopag and thrombopoietin on in vitro platelet function. Exp Hematol. 2009;37:1030–7.
Harker LA, Hunt P, Marzec UM, et al. Regulation of platelet production and function by megakaryocyte growth and development factor in nonhuman primates. Blood. 1996;87:1833–44.
Jenkins JM, Williams D, Deng Y, et al. Phase 1 clinical study of eltrombopag, an oral, nonpeptide thrombopoietin receptor agonist. Blood. 2007;109:4739–41.
Promacta prescribing information: GlaxoSmithKline, Inc; 2008.
Saleh MN, Bussel JB, Cheng G, et al. Safety and efficacy of eltrombopag for treatment of chronic immune thrombocytopenia: results of the long-term, open-label EXTEND study. Blood. 2013;121:537–45.
Kuter DJ, Mathias SD, Rummel M, et al. Health-related quality of life in nonsplenectomized immune thrombocytopenia patients receiving romiplostim or medical standard of care. Am J Hematol. 2012;87:558–61.
Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet. 2011;377:393–402.
Bussel JB, Buchanan GR, Nugent DJ, et al. A randomized, double-blind study of romiplostim to determine its safety and efficacy in children with immune thrombocytopenia. Blood. 2011;118:28–36.
McHutchison JG, Dusheiko G, Shiffman ML, et al. Eltrombopag for thrombocytopenia in patients with cirrhosis associated with hepatitis C. N Engl J Med. 2007;357:2227–36.
Olnes MJ, Scheinberg P, Calvo KR, et al. Eltrombopag and improved hematopoiesis in refractory aplastic anemia. N Engl J Med. 2012;367:11–9.
Pecci A, Panza E, Pujol-Moix N, et al. Position of nonmuscle myosin heavy chain IIA (NMMHC-IIA) mutations predicts the natural history of MYH9-related disease. Hum Mutat. 2008;29:409–17.
Pecci A, Gresele P, Klersy C, et al. Eltrombopag for the treatment of the inherited thrombocytopenia deriving from MYH9 mutations. Blood. 2010;116:5832–7.
Zimrin A, Chumsri S. Profound thrombocytopenia in patient with ITP treated with chemotherapy for breast cancer and subsequent remission after romiplostim. Ann Hematol. 2013 [Epub ahead of print].
Demeter J, Istenes I, Fodor A, et al. Efficacy of romiplostim in the treatment of chemotherapy induced thrombocytopenia (CIT) in a patient with mantle cell lymphoma. Pathol Oncol Res. 2011;17:141–3.
Parameswaran R, Soff G. Romiplostim in a case series of chemotherapy-induced thrombocytopenia. Blood. 2011;118:1170.
Winer E, Safran H, Karaszewska B, et al. Safety and efficacy of eltrombopag (epag) versus placebo (pbo) for the treatment (tx) of chemotherapy-induced thrombocytopenia (CIT) in patients with solid tumors receiving gemcitabine (gem)-based chemotherapy (ctx): a phase I study. J Clin Onc. 2012;30:9117.
Douglas VK, Tallman MS, Cripe LD, Peterson LC. Thrombopoietin administered during induction chemotherapy to patients with acute myeloid leukemia induces transient morphologic changes that may resemble chronic myeloproliferative disorders. Am J Clin Pathol. 2002;117:844–50.
Kuter DJ, Mufti GJ, Bain BJ, Hasserjian RP, Davis W, Rutstein M. Evaluation of bone marrow reticulin formation in chronic immune thrombocytopenia patients treated with romiplostim. Blood. 2009;114:3748–56.
Rodeghiero F, George J, Rummel M, et al. Results from a phase IV open-label study evaluating changes in bone marrow morphology in adult immune thrombocytopenia (ITP) patients receiving romiplostim: analysis of the 1-year romiplostim cohort. Haematologia (Budap). 2012;97:611.
Brynes R, Orazi A, Verma S, Brainsky A, Bailey C, Bakshi K. Evaluation of bone marrow reticulin in patients with chronic immune thrombocytopenic purpura (ITP) treated with eltrombopag—data from the EXTEND Study. Blood. 2011;118:528.
Brynes R, Orazi A, Wong R, Bakshi K, Bailey C, Brainsky A. A longitudinal prospective study evaluating the effects of eltrombopag treatment on bone marrow in patients with chronic immune thrombocytopenia: interim analysis at 1 year. Blood. 2012;120.
Giagounidis A, Mufti G, Kantarjian H, et al. Treatment with the thrombopoietin (TPO)-receptor agonist romiplostim in thrombocytopenic patients (pts) with low or intermediate-1 (Int-1) risk myelodysplastic syndrome (MDS): results of a randomized, double-blind, placebo(pbo)-controlled study. Blood. 2011;118:117.
Sarpatwari A, Bennett D, Logie JW, et al. Thromboembolic events among adult patients with primary immune thrombocytopenia in the United Kingdom General Practice Research Database. Haematologica. 2010;95:1167–75.
Bennett I, Forssen U, Enger V, Nelson J. Risk of thrombotic events among patients with chronic idiopathic thrombocytopenia purpura (ITP). Haematologia (Budap). 2008;93(s1):125.
Severinsen MT, Engebjerg MC, Farkas DK, et al. Risk of venous thromboembolism in patients with primary chronic immune thrombocytopenia: a Danish population-based cohort study. Br J Haematol. 2011;152:360–2.
Afdhal NH, Giannini EG, Tayyab G, et al. Eltrombopag before procedures in patients with cirrhosis and thrombocytopenia. N Engl J Med. 2012;367:716–24.
Ghilardi N, Wiestner A, Kikuchi M, Ohsaka A, Skoda RC. Hereditary thrombocythaemia in a Japanese family is caused by a novel point mutation in the thrombopoietin gene. Br J Haematol. 1999;107:310–6.
Wiestner A, Padosch SA, Ghilardi N, et al. Hereditary thrombocythaemia is a genetically heterogeneous disorder: exclusion of TPO and MPL in two families with hereditary thrombocythaemia. Br J Haematol. 2000;110:104–9.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Kuter, D.J. The biology of thrombopoietin and thrombopoietin receptor agonists. Int J Hematol 98, 10–23 (2013). https://doi.org/10.1007/s12185-013-1382-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-013-1382-0