
Abstract
Invariant natural killer T (iNKT) cells are characterized by the expression of an invariant Vα14–Jα18 paired with Vβ8/7/2 in mice, and Vα24–Jα18 with Vβ11 in humans, that recognizes glycolipids, such as α-galactosylceramide (α-GalCer), presented on the MHC class I-like molecule, CD1d. iNKT cells act as innate T lymphocytes and serve as a bridge between the innate and acquired immune systems. iNKT cells augment anti-tumor responses by producing IFN-γ, which acts on NK cells to eliminate MHC-non-restricted (MHC−) target tumor cells, and on CD8+ cytotoxic T lymphocytes to directly kill MHC-restricted (MHC+) tumor cells. Thus, when iNKT cells are activated by α-GalCer-pulsed dendritic cells, both MHC− and MHC+ tumor cells can be effectively eliminated. Both of these tumor cell types are simultaneously present in cancer patients, and at present iNKT cells are only the cell type capable of eliminating them. Based on these findings, we have developed iNKT cell-targeted adjuvant immunotherapies with strong anti-tumor activity in humans. However, two-thirds of patients were ineligible for this therapy due to the limited numbers of iNKT cells in their bodies. In order to overcome the problem in cancer patients, we successfully established a method to generate iNKT cells with adjuvant activity from embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs). In this review, we would like to outline the clinical potential for iNKT cells derived from ESCs and iPSCs for cancer immunotherapy, and the technical hurdles that must be overcome if we achieve effective ESC/iPSC-mediated cancer therapies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Invariant natural killer T (iNKT) cells are characterized by the expression of a unique single invariant antigen receptor encoded by Vα14–Jα18, mainly associated with Vβ8.2 in mice [1] and Vα24–Jα18 with Vβ11 in humans [2]. In addition, the limited repertoire of iNKT cells appears to be autoreactive and, as a result, these cells are persistently activated by endogenous glycolipid antigens in association with the monomorphic MHC class I-like molecule, CD1d [3–5]. These characteristics are quite distinct from conventional T cells, which have a highly diverse repertoire and mainly recognize peptide antigens with polymorphic MHC molecules. The invariant Vα14–Jα18 TCRα is used only by iNKT cells, and not by conventional T cells. Thus, when a pre-rearranged Vα14–Jα18/Vβ8.2 gene is introduced into recombination activating gene (RAG)-deficient mice, only iNKT cells (and not conventional T cells or NK cells) develop, defining iNKT as a unique lymphocyte subset [6]. Conversely, Jα18 knockout mice specifically lack iNKT cells, but have normal numbers of conventional T cells [7].
Here, we describe the properties of iNKT cells and their adjuvant activity, features that are now being manipulated therapeutically to treat human cancer. We also describe a method for the generation of large numbers of iNKT cells from ES cells or iPS cells in vitro, which is an important advance in overcoming the problem of the limited number of iNKT cells in advanced cancer patients.
Mechanisms of iNKT cell-mediated adjuvant activity augmenting anti-tumor responses
The promiscuous production of T helper cell (Th)1, Th2, and Th17 cytokines is a characteristic feature of iNKT cells [8–10]. Their recognition of self-ligands leads to the accumulation of cytokine mRNAs and up-regulation of activation markers, indicating that iNKT cells in the steady state are already primed and ready to quickly mediate their effector functions, serving as a bridge between innate and acquired immunity [1]. However, their recognition of endogenous ligands does not elicit cytokine production, only transcript accumulation, as iNKT cells require additional signals to produce cytokines mediating their functions.
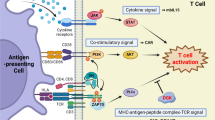
The nature of these additional signals is the key event that determines iNKT cell function. Immature dendritic cells (DCs) in the steady state can capture antigens, but are unable to activate conventional T cells. However, iNKT cells can be activated by immature DCs and then reciprocally induce the maturation of DCs. A single in vivo injection of α-galactosylceramide (α-GalCer), a synthetic exogenous glycolipid ligand for iNKT cells presented by CD1d [6], induces a burst of interleukin (IL)-12 production (at 6 h) by DCs followed by Th1 cytokine interferon (IFN)-γ production by iNKT cells (at 24 h) [11–18]. This response is triggered by interaction between CD40 on DCs and CD40 ligand (CD40L) on iNKT cells, which occurs within 2–6 h after α-GalCer injection [19]. The iNKT cells are necessary for this DC maturation, as it does not occur in Jα18 −/− iNKT cell-deficient mice. The maturation of DCs is blocked in tumor patients, due to their production of immunosuppressive cytokines, such as TGF-β and IL-10 [20, 21]. Thus, the in vivo maturation of DCs by activated iNKT cells is an important strategy for the augmentation of protective immunity in cancer patients.
Because of their self-reactivity and ability to quickly release large amounts of cytokines, iNKT cells link the two immune systems, serving as a bridge between the innate and acquired systems, and thus augment protective immune responses, including anti-tumor immune responses, through the activation of NK and CD8+ CTLs [9]. This augmentation of protective immunity is known as the iNKT cell-mediated adjuvant effect, and is critical for tumor eradication. Since tumor cells do not provide any adjuvant effects or “danger signals” to activate the immune system, they fail to induce tumor-specific immune responses sufficiently potent to eradicate tumor cells even when tumor-specific T cells are present. Moreover, there are two general types of tumor cells in a tumor mass: one is MHC+ and the other is MHC− [20, 21], and effective tumor immunity requires that both types of tumor cells be eliminated simultaneously. iNKT cells are the only cell type that is able both to interact with immature DCs, inducing their maturation, and to augment the function of both NK cells and CD8+ CTLs.
IFN-γ produced by activated iNKT cells is a key cytokine for mediating adjuvant activity. By this mechanism, iNKT cells can induce the maturation of DCs, which thereby acquire the ability to present tumor antigens to CTLs. The activated CTLs can then eliminate MHC+ tumor cells. IFN-γ can also activate NK cells, which kill MHC− tumor targets. The efficacy of this approach has been demonstrated in several studies in which treatment of tumor-bearing mice with α-GalCer-pulsed DCs to activate endogenous iNKT cells leads to the eradication of established metastatic tumors [22]. Thus, the activation of endogenous iNKT cells is a promising strategy for the treatment of cancer by selectively triggering protective anti-tumor immunity.
iNKT cell-targeted adjuvant cell therapy for patients with advanced lung cancer
Based on the above translational studies in tumor-bearing mice and the highly conserved nature of the iNKT/CD1d system, α-GalCer was approved for use as a drug for clinical applications. Several clinical trials involving the injection of α-GalCer-pulsed DCs have been carried out in patients with cancer, including colon cancer, multiple myeloma, anal cancer, and renal cell cancer [23, 24]. Although no clear tumor reduction was detected, tumor markers were significantly decreased.
We and colleagues have completed a phase I/IIa clinical trial of iNKT cell-targeted adjuvant therapy using α-GalCer-pulsed DCs (total 4 × 109 cells per patient in four consecutive injections at 1 week intervals) on 17 patients with advanced lung cancer, including stage IV, IIIB primary cancer, and recurrent tumor after surgery [25–28]. The patients’ peripheral blood mononuclear cells were cultured with GMP grade GM-CSF and IL-2 for 2 weeks to increase the number of DCs, pulsed with α-GalCer for 24 h, and then administered intravenously into the donor. Significant increases in the number of IFN-γ-producing cells were detected in 60 % of enrolled patients (10 of 17) and this correlated with a prolonged median survival time (MST) of 31.9 months without tumor progression and metastasis, with only a primary treatment and no further additional treatment. Interestingly, none of the 10 patients with longer survival times showed tumor regression [28]. By contrast, the patient group with low IFN-γ production had a MST of 9.7 months, which is equivalent to the MST of 10 months after treatment with commercially available molecular target drugs such as anti-VEGFR antibody (Avastin: 13.1 months), anti-EGFR (Erbitux: 10.1 months), Folic acid inhibitor (Alimta: 8.3 months), EGFR inhibitor (Iressa/Tarceva: 6.7 months). Thus, IFN-γ appears to be a good biological marker predictive of a favorable clinical course.
However, despite the clear anti-tumor activity of the iNKT cell-targeted therapy, two-thirds of patients were not eligible, as they no longer had sufficient iNKT cells (<10 cells/ml of blood).
Generation of ES cells and iPS cells from mature iNKT cells
In order to overcome the problem of the limited number of iNKT cells in cancer patients, we attempted to establish methods to supply iNKT cells. Embryonic stem (ES) cells are a powerful model system to study in vitro lymphocyte differentiation, addressing the questions on the cells with iNKT precursor potential and the ability of iNKT cells to produce both Th1 and Th2 cytokines during their development. For example, embryoid bodies generated from ES cells contain CD34+ cells that develop into lymphocytes when co-cultured with OP9 stromal cells plus appropriate cytokines [29]. However, in most cases, ES cells generate B and NK cells, but not T or iNKT cells on OP9 co-culture [30, 31]. In contrast, OP9 stromal cells transduced with Notch ligand delta-like1 (OP9/Dll-1) are used for the directed differentiation of ES cells to T cell lineages. Over-expression of active Notch1 directs stem cells to differentiate into immature DP T cells and inhibits B cell development, indicating that Notch signaling is required as a proximal event in T cell commitment from progenitors [32, 33].
Another approach is to use cloned ES (iNKT–ES) cells established by nuclear transfer of a cell with a rearranged T cell receptor (Tcr) gene [34, 35]. Interestingly, iNKT cells retain high genome re-programmability (71 % for iNKT vs. 12 % for T) [35]. The iNKT cell nucleus may therefore provide important clues for analysis of differentiation pathways from ES cells to mature iNKT cells. Based on these findings, we have established iNKT–ES cells generated by nuclear transfer of C57BL/6 (B6) liver iNKT cells, into B6D2F1 unfertilized eggs followed by re-transfer of 2-cell stage nuclei into the 2-cell stage cytoplasm of (B6D2F1xICR)F1 in vivo fertilized embryos. In total, six blastocytes were obtained from the 147 initial 2-cell stage embryos. This efficiency (4 %) is similar to that reported for clones from cumulus cells (2.3–6.9 %) or adult fibroblasts (1.1–3.8 %) [35]. Four of six cell lines established were analyzed by genomic PCR using primers that detect the non-rearranged Vα14 gene and primers that specifically detect the Vα14–Jα18 gene rearrangement. This analysis demonstrated that all established iNKT–ES clones contained the rearranged genomic Vα14–Jα18 on one chromosome and the germline configuration on the other [36].
Since iPS cells are functionally equivalent to ES cells in many respects [37–39], these results suggested the feasibility of using iPS cells for iNKT cell-targeted adjuvant therapy. It is important to note that iPS cells rather than ES cells are more feasible in the clinical setting, as embryos or donor oocytes are unnecessary for the generation of iPS cells. In addition, iPS cells are fully syngeneic, while ES cells, even if introduced with patient cell nucleus, carrying oocyte-derived mitochondrial maternal antigens may trigger allogeneic immune responses.
Although several attempts have been made to generate iPS cells from mature B and T cells by the introduction of Oct3/4, Sox2, Klf4 and c-Myc, the efficiency is very low [40]. Since iNKT cells have a higher reprogramming activity compared to conventional T cells, we thought that iNKT cells would be suitable for generating iPS cells. In fact, we successfully generated iPS cells from mature iNKT cells and were then able to generate functional iNKT cells from the iPS cells (iNKT–iPS) in vitro. iNKT cells (106) isolated from spleen cells of normal B6 or B6 iNKT clone mice were first activated by anti-CD3/CD28 together with IL-12 and IL-2 for a week, followed by reprogramming according to the conventional protocol [37]. We established three independent iPS cell lines, which were iNKT cell-derived, based on the presence of rearranged Vα14Jα18 sequences. The expression of SSEA1, Oct3/4 and Nanog, and also that of endogenous Oct3/4, Sox2, Klf4, Nanog, Ecat1, Gdf3, Rex1 and Zfp296 mRNA in the iPS cells were detected at similar levels to those observed in ES cells. Genome-wide gene expression profiles of the iPS cell lines showed a higher correlation coefficient to ES cells than to splenic iNKT cells [41].
Generation of functional iNKT cells from ES cells and iPS cells
Using iNKT–ES cells, we established an iNKT–ES culture system using OP9/Dll-1 as an adherent cell layer in the presence of IL-7 and Flt-3 ligand (Flt3L). In the T cell development, pre-TCR signaling is essential for differentiation of CD4−CD8− double negative (DN) cells to the DP stage. The DN cells are divided into DN1–DN4 subsets, according to the expression of CD44 and CD25. The earliest CD44+CD25− (DN1) cells provide the CD44+CD25+ (DN2) population that progress to the CD44−CD25+ (DN3) stage, followed by the CD44−CD25− (DN4) stage. iNKT–ES cells were sustained in culture as the DN1 population from days 11 to 14. On the following days of culture, cells displayed the phenotypes similar to thymic DN2/3 population (CD44lo/−CD25hi) by day 16, subsequently into DN3/4 cells (CD44− CD25−/int) on day 17. Finally, DP cells appeared on day 20 of culture, most of which α-GalCer/CD1d dimer+/TCRβ+ iNKT cells. Therefore, it seems that the iNKT–ES culture system populated iNKT cells in vitro, tracing the similar developmental progression detected in the un-manipulated ES culture system as well as thymocyte in vivo [36].
Although there are no reports of cytokine production of DP iNKT cells, ES-derived iNKT cells produce IL-4, but only low levels of IFN-γ. The results indicate that they are functionally equivalent to IL-4-producing iNKT cells of the NK1.1−CD4+ or CD4−CD8− iNKT cells in the thymus that mainly produce IL-4 [42, 43]. Although there is no report on cytokine production of DP iNKT cells, it is likely that IL-4-producing iNKT cells developed from iNKT–ES cells are similar to CD24hi DP iNKT cells in the thymus, which give rise to NK1.1−CD4+ or CD4−CD8− iNKT cells. Moreover, in keeping with their immaturity, these IL-4-producing iNKT cells showed potent proliferative activity [36].
Even though there are no reports of the successful regeneration of functional B or T cells from lymphocyte-derived iPS cells [44–48], the potential of the iNKT–iPS cell lines to re-differentiate into mature functional iNKT cells in vitro was investigated using a modified above protocol [36, 49] of a 20- to 25-day culture system containing IL-7 and Flt3L as supportive cytokines and OP9/Dll-1 as stromal cells [50].
We first attempted to investigate the potential of iNKT cell development from iNKT–iPS cells using the 20-day culture system used for analysis of ES cells described above. Similar to ES-derived iNKT cells, iPS-derived iNKT cells mainly produced IL-4 on OP9/Dll-1 culture for 20 days, indicating that iNKT–iPS cells are potentially similar to iNKT–ES cells.
It is important to obtain high yield of iNKT cells with desired function for the establishment of iNKT cell therapy. For this purpose, we attempted to develop a new 25-day culture system efficiently to generate functional iNKT cells from the iNKT–iPS cells in vitro. The addition of IL-7 at the culture day 20 for 5 days yielded the highest numbers of iNKT cells, and also the highest amounts of IFN-γ production, equivalent to those of splenic iNKT cells. Under these conditions, 1 × 105 iPS cells gave rise to 3 × 107 iPS-derived iNKT cells at the culture day 25 that expressed TCRβ on their surface and bound α-GalCer-loaded soluble CD1d dimer, which are specifically recognized by the iNKT-specific TCR. The iPS-derived iNKT cells did not express NK1.1. Moreover, a significant fraction of the iPS-derived iNKT cells were doubly positive for CD4 and CD8. Therefore, the iPS-derived iNKT cells generated in this 25-day culture system were similar to immature DP thymocytes than mature liver iNKT cells. Moreover, they were CD24hi, CD44lo/int, CD62Lhi, CD69+, CD122lo/− and NKG2D−, which are phenotypically similar to the earliest thymic iNKT cells, so-called stage 0 iNKT cells.
In vivo function of iPS-derived iNKT cells
The in vivo survival and function of iPS-derived iNKT cells were investigated after transfer into iNKT cell-deficient (Jα18 −/−) mice [50]. Similar to the ability of normal wild type mature iNKT cells to repopulate the Jα18 −/− mice, a considerable number of iPS-derived iNKT cells could repopulate in the recipient liver 2 weeks after adoptive transfer. Although the in vitro-generated iPS-derived iNKT cells that we injected had an immature phenotype (see previous section), the iNKT cells recovered from the liver were CD24lo, CD44hi, CD62Llo, CD69+, CD122+, and NKG2D+, phenotypically similar to mature liver iNKT cells in wild type mice, indicating that their maturation occurs in vivo. Similar to the in vitro activity of iPS-derived iNKT cells obtained in the 25-day cultures, the iPS-derived iNKT cells transferred in vivo produced large amounts of IFN-γ upon antigen stimulation with α-GalCer intravenously, and expanded significantly in number (~10-fold increase: 0.3–0.4 % to 4.9–5.7 %). These iPS-derived iNKT cells also showed down-modulated TCRβ expression, consistent with their in vivo activation. Similar results were observed after transferring ES-derived iNKT cells, indicating both ES- and iPS-derived iNKT cells survive and mature after transfer in vivo.
Adjuvant activity of ES and iPS-derived iNKT cells
As the iPS-derived iNKT cells produced IFN-γ upon stimulation with α-GalCer (see “Generation of functional iNKT cells from ES cells and iPS cells”), we investigated their adjuvant effects on anti-tumor responses in mice. The Jα18 −/− mice that had received iPS-derived iNKT cells (4 × 106) were immunized with cell-associated ovalbumin (OVA) (spleen cells from Tap −/− mice that were treated with hypertonic medium in the presence of OVA) together with α-GalCer (TOG). A week later, the mice were challenged with OVA257–264 peptide and analyzed for IFN-γ production by OVA-specific CTLs and by NK cells [50]. We observed significant production of IFN-γ by the NK cells and OVA-specific CTLs, as well as an increase in the number of these CTLs (>70 times higher than the control group). Thus, iPS-derived iNKT cells were shown to function as a cellular adjuvant for both innate and adaptive immune responses.
Under these conditions, iPS-derived iNKT cells significantly augmented antigen-specific anti-tumor CTL responses against EL-4-derived OVA-bearing EG7 tumor cells (OVA serves as tumor antigen in this system) but not against EL4 in TOG-immunized B6 mice. The growth of OVA-bearing EG7 tumor cells in Jα18 −/− mice was significantly suppressed by adoptive transfer of iPS-derived iNKT cells, whereas the growth of EL4 was not. Therefore, the iPS-derived iNKT cells are functionally competent to mediate adjuvant activity in vivo.
Towards generation of human iPSCs that give rise to iNKT cells in vitro
These studies using mouse ESCs and iPSCs and the functional similarity of human iNKT cells with those in mice point to the clinical potential of human ESCs or iPSCs that harbor rearranged Tcra and Tcrb genes specific for iNKT cells for cancer treatment. To test this possibility, we need to establish human ESCs or iPSCs able to give rise iNKT cells in vitro and also optimize induction assays for functionally maturated T cells from human ESCs or iPSCs. Since generation of human ESCs with rearranged Tcr genes needs nuclear transfer of T cell nuclei into human unfertilized eggs, use of iPSCs is more realistic option at this point. We, however, found it is not simple to induce iPSCs from human iNKT cells circulating in the peripheral blood due to two main technical hurdles: the low frequency of iNKT cells in the peripheral blood, and the low induction efficacy of iPSCs from adult mature T cells. In our pilot experiments, we observed that iNKT cells represent 0.0001–1 % of mononuclear cells in peripheral blood, and that this frequency is quite variable in human population. This suggests we can collect approximately 102–106 iNKT cells from 200 ml of adult peripheral blood and this number theoretically warrants us to generate iPSCs from peripheral blood lymphocytes (PBL) since previous studies reported reprogramming frequency of peripheral blood T cells was approximately 0.1 % (50 hESC-like colonies were observed from 5 × 104 peripheral blood T cells) by Sendai virus vector (SeV)-mediated expression of OCT3/4, SOX2, KLF4 and c-MYC (OSKM) [51]. Moreover, we found co-expression of SV40 T-antigen with OSKM further increased reprogramming rate of cord blood T cells to approximately 0.5 % (474 hESC-like colonies were observed from 1 × 105 cord blood T cells). In fact, we have successfully induced ample iPSCs, which were shown to possess productively rearranged Tcra and Tcrb genes (unpublished), from CD4+ and CD8+ cord blood T lymphocytes. We, however, found the reprogramming rates of adult PBL T cells were more than 102 lower than those of cord blood T cells. This number is insufficient to induce iNKT–iPSCs from PBL containing average number of iNKT cells using currently available technology. We clearly need further technical advances to improve reprogramming frequency of adult T cells.
An alternative strategy to generate human T-iPSCs harboring rearranged Tcra and Tcrb gene configurations specific for iNKT cells may involve induced replacement of rearranged variable regions in T-iPSCs with those in iNKTs using zinc finger nuclease (ZFN) or transcription activator-like effector nuclease (TALEN) technology. Recently, several groups have reported gene targeting in human iPSCs by homologous recombination using these technologies. We need two consecutive steps for this replacement at both Tcra and Tcrb genes, as the targeting reaction by either ZFNs or TALEN does not allow efficient direct replacement. Therefore, first steps should be deletion of genomic regions encompassing rearranged variable region of Tcra or Tcrb loci by ZFNs or TALEN. Indeed, ZFNs have been shown to efficiently produce targeted insertion of small genetic elements concomitant with genomic deletions [52, 53]. We may thus induce deletion of Tcra and Tcrb genomic regions and concurrently introduce some known ZFN- or TALEN-target sequence, which is not present on the human genome, to enable insertion of DNA fragment encoding other rearranged Tcra or Tcrb region by secondary targeting reaction (Fig. 1a). These T-iPSCs with deletion of variable regions at Tcra and Tcrb genes could be versatile recipient for exogenous variable region genes in the second step. Flanking genomic sequences of this ZFN- or TALEN-target sequence are used to generate donor vectors containing drug selection markers to knock-in NKT cell-specific variable regions. Donor vectors are transfected into versatile recipient iPSCs, together with ZFN or TALEN. After cloning of homologous recombinants, we may delete selection marker genes using PiggyBac or Cre-loxP system (Fig. 1b). Moreover, this technology is theoretically applicable to express antigen-specific T cell receptors once we could know their primary structures.

Strategy for iNKT-specific TCR genome editing in human T-iPSC. a Generation of T-iPSC harboring TCR knock-in cassette (mROSA26) using ssDNA oligos together with ZFNs or TALEN introduction. b Generation of T-iPSC harboring NKT cell-specific variable regions using TCR knock-in cassette-specific ZFNs (TALEN). After cloning, selection marker is deleted by PiggyBac or Cre-lox. By way of example, iNKT TCRα is shown in this figure
Although human iPSCs that can give rise to iNKT cells may be an attractive device to realize iNKT cell-mediated immunotherapy for cancer treatment, above considerations indicate that we still need technical development even at the first step. Moreover, functional properties of iPSCs are known to be quite variable, but we still do not have legitimate quality and safety standards for human T-iPSCs. Although differentiation protocols for the induction of T cells from ESCs or iPSCs are not fully optimized, we can check differentiation ability of these cells in vitro and tumorigenicity of induced cells in immunocompromised mice. As a first step to develop iPSC-mediated immunotherapy, we should perform small-scale banking of human T-iPSCs, in which we describe nucleotide sequences of Tcr genes, HLA haplotype, differentiation ability in vitro, tumorigenicity in mice and possibly other parameters as a pilot trial. On one hand, this bank should be publically shared and enabled for access of researchers harboring various ideas. On the other hand, we need to select well-qualified T-iPSCs to generate versatile recipient iPSCs for further modifications of Tcr loci.
Future perspectives
Our previous studies in mice have clearly provided a proof of concept for the use of iPSC-derived iNKT cells for adjuvant cell therapy against cancer, which is composed of four segments: (1) collection of iNKT cells, (2) reprogramming of iNKT cells into iPSCs, (3) re-differentiation of iNKT cell-derived iPSCs into iNKT cells and their expansion in vitro, and (4) injection of iPSC-derived iNKT cells into tumor-bearing animals (Fig. 2). Since the functions and α-GalCer reactivity of iNKT cells are highly conserved between mice and humans, and human CD1 is monomorphic as well as murine CD1d, it is conceptually possible to tailor iNKT cell-targeted adjuvant cell therapy to human cancer treatment beyond extensive heterogeneity of human HLA. Establishment of iPSC lines that can differentiate into functionally competent iNKT cells should be addressed to test the clinical reality for iPSC-derived iNKT cell-mediated immunotherapy.

Schematic representation of iNKT cell supplemental cellular immunotherapy. iPS cells that differentiate into iNKT cells can be developed from peripheral mature iNKT cells by the introduction of Oct3/4, Sox2, Klf4 and c-Myc (OSKM) or iNKT-specific TCR genome editing shown in Fig. 1. The iPSCs can be saved for later use, and differentiated into iNKT cells with desired properties using this reliable culture system. iNKT cell-mediated cellular immunotherapy shows promising anti-tumor activity, and it will be crucial to establish methods for delivering iNKT cells to the body
References
Taniguchi M, Harada M, Kojo S, et al. The regulatory role of Vα14 NKT cells in innate and acquired immune response. Annu Rev Immunol. 2003;21:483–513.
Lantz O, Bendelac A. An invariant T cell receptor α chain is used by a unique subset of major histocompatibility complex class I-specific CD4+ and CD4−8− T cells in mice and humans. J Exp Med. 1994;180:1097–106.
Bendelac A. Positive selection of mouse NK1+ T cells by CD1-expressing cortical thymocytes. J Exp Med. 1995;182:2091–6.
Bendelac A, Lantz O, Quimby ME, et al. CD1 recognition by mouse NK1+ T lymphocytes. Science. 1995;268:863–5.
Exley M, Garcia J, Balk SP, et al. Requirements for CD1d recognition by human invariant Vα24+CD4−CD8− T cells. J Exp Med. 1997;186:109–20.
Kawano T, Cui J, Koezuka Y, et al. CD1d-restricted and TCR-mediated activation of Valpha14 NKT cells by glycosylceramides. Science. 1997;278:1626–9.
Cui J, Shin T, Kawano T, et al. Requirement for Vα14 NKT cells in IL-12-mediated rejection of tumors. Science. 1997;278:1623–6.
Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol. 2007;25:297–336.
Taniguchi M, Seino K, Nakayama T. The NKT cell system: bridging innate and acquired immunity. Nat Immunol. 2003;4:1164–5.
Michel ML, Keller AC, Paget C, et al. Identification of an IL-17-producing NK1.1neg iNKT cell population involved in airway neutrophilia. J Exp Med. 2007;204:995–1001.
Tomura M, Yu WG, Ahn HJ, et al. A novel function of Vα14+CD4+ NKT cells: stimulation of IL-12 production by antigen-presenting cells in the innate immune system. J Immunol. 1999;163:93–101.
Kitamura H, Iwakabe K, Yahata T, et al. The natural killer T (NKT) cell ligand alpha-galactosylceramide demonstrates its immunopotentiating effect by inducing interleukin (IL)-12 production by dendritic cells and IL-12 receptor expression on NKT cells. J Exp Med. 1999;189:1121–8.
Gonzalez-Aseguinolaza G, de Oliveira C, Tomaska M, et al. α-Galactosylceramide-activated Vα14 natural killer T cells mediate protection against murine malaria. Proc Natl Acad Sci USA. 2000;97:8461–6.
Trobonjaca Z, Leithäuser F, Möller P, et al. Activating immunity in the liver. I. Liver dendritic cells (but not hepatocytes) are potent activators of IFN-γ release by liver NKT cells. J Immunol. 2001;167:1413–22.
Stober D, Jomantaite I, Schirmbeck R, et al. NKT cells provide help for dendritic cell-dependent priming of MHC class I-restricted CD8+ T cells in vivo. J Immunol. 2003;170:2540–8.
Hermans IF, Silk JD, Gileadi U, et al. NKT cells enhance CD4+ and CD8+ T cell responses to soluble antigen in vivo through direct interaction with dendritic cells. J Immunol. 2003;171:5140–7.
Fujii S, Shimizu K, Smith C, et al. Activation of natural killer T cells by α-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J Exp Med. 2003;198:267–79.
Fujii S, Liu K, Smith C, et al. The linkage of innate to adaptive immunity via maturing dendritic cells in vivo requires CD40 ligation in addition to antigen presentation and CD80/86 costimulation. J Exp Med. 2004;199:1607–18.
Fujii S, Shimizu K, Hemmi H, et al. Innate Vα14+ natural killer T cells mature dendritic cells, leading to strong adaptive immunity. Immunol Rev. 2007;220:183–98.
Khong HT, Restifo NP. Natural selection of tumor variants in the generation of tumor escape phenotypes. Nat Immunol. 2002;3:999–1005.
Yang L, Carbone DP. Tumor-host immune interactions and dendritic cell dysfunction. Adv Cancer Res. 2004;92:13–27.
Toura I, Kawano T, Akutsu Y, et al. Cutting edge: inhibition of experimental tumor metastasis by dendritic cells pulsed with a-galactosylceramide. J Immunol. 1999;163:2387–91.
Nieda M, Okai M, Tazbirkova A, et al. Therapeutic activation of Vα24+Vβ11+ NKT cells in human subjects results in highly coordinated secondary activation of acquired and innate immunity. Blood. 2004;103:383–9.
Chang DH, Osman K, Connolly J, et al. Sustained expansion of NKT cells and antigen-specific T cells after injection of α-galactosyl-ceramide loaded mature dendritic cells in cancer patients. J Exp Med. 2005;201:1503–17.
Ishikawa A, Motohashi S, Ishikawa E, et al. A phase I study of α-galactosylceramide (KRN7000)-pulsed dendritic cells in patients with advanced and recurrent non-small cell lung cancer. Clin Cancer Res. 2005;11:1910–7.
Motohashi S, Ishikawa A, Ishikawa E, et al. A phase I study of in vitro expanded natural killer T cells in patients with advanced and recurrent non-small cell lung cancer. Clin Cancer Res. 2006;12:6079–86.
Motohashi S, Nakayama T. Clinical applications of natural killer T cell-based immunotherapy for cancer. Cancer Sci. 2008;99:638–45.
Motohashi S, Nagato K, Kunii N, et al. A phase I–II study of α-galactosylceramide-pulsed IL-2/GM-CSF-cultured peripheral blood mononuclear cells in patients with advanced and recurrent non-small cell lung cancer. J Immunol. 2009;182:2492–501.
Nakayama N, Fang I, Elliott G. Natural killer and B-lymphoid potential in CD34+ cells derived from embryonic stem cells differentiated in the presence of vascular endothelial growth factor. Blood. 1998;91:2283–95.
Nakano T, Kodama H, Honjo T. Generation of lymphohematopoietic cells from embryonic stem cells in culture. Science. 1994;265:1098–101.
Cho SK, Webber TD, Carlyle JR, et al. Functional characterization of B lymphocytes generated in vitro from embryonic stem cells. Proc Natl Acad Sci USA. 1999;96:9797–802.
de Pooter RF, Cho SK, Carlyle JR, et al. In vitro generation of T lymphocytes from embryonic stem cell-derived prehematopoietic progenitors. Blood. 2003;102:1649–53.
Schmitt TM, de Pooter RF, Gronski MA, et al. Induction of T cell development and establishment of T cell competence from embryonic stem cells differentiated in vitro. Nat Immunol. 2004;5:410–7.
Hochedlinger K, Jaenisch R. Monoclonal mice generated by nuclear transfer from mature B and T donor cells. Nature. 2002;415:1035–8.
Inoue K, Wakao H, Ogonuki N, et al. Generation of cloned mice by direct nuclear transfer from natural killer T cells. Curr Biol. 2005;15:1114–8.
Watarai H, Rybouchkin A, Hongo N, et al. Generation of functional NKT cells in vitro from embryonic stem cells bearing rearranged invariant Vα14–Jα18 TCRα gene. Blood. 2010;115:230–7.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76.
Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–7.
Wernig M, Meissner A, Foreman R, et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007;448:318–24.
Hong H, Takahashi K, Ichisaka T, et al. Suppression of induced pluripotent stem cell generation by the p53–p21 pathway. Nature. 2009;460:1132–5.
Schmitt TM, Zúñiga-Pflücker JC. Induction of T cell development from hematopoietic progenitor cells by delta-like-1 in vitro. Immunity. 2002;17:749–56.
Benlagha K, Kyin T, Beavis A, et al. A thymic precursor to the NK T cell lineage. Science. 2002;296:553–5.
Pellicci DG, Hammond KJ, Uldrich AP, et al. A natural killer T (NKT) cell developmental pathway involving a thymus-dependent NK1.1(−)CD4(+) CD1d-dependent precursor stage. J. J Exp Med. 2002;195:835–44.
Hanna J, Markoulaki S, Schorderet P, et al. Direct reprogramming of terminally differentiated mature B lymphocytes to pluripotency. Cell. 2008;133:250–64.
Brown M, Rondon E, Rajesh D, et al. Derivation of induced pluripotent stem cells from human peripheral blood T lymphocytes. PLoS One. 2010;5:e11373.
Seki T, Yuasa S, Oda M, et al. Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell. 2010;7:11–4.
Loh YH, Hartung O, Li H, et al. Reprogramming of T cells from human peripheral blood. Cell Stem Cell. 2010;7:15–9.
Staerk J, Dawlaty MM, Gao Q, et al. Reprogramming of human peripheral blood cells to induced pluripotent stem cells. Cell Stem Cell. 2010;7:20–4.
Watarai H, Nakagawa R, Omori-Miyake M, et al. Methods for detection, isolation and culture of mouse and human invariant NKT cells. Nat Protoc. 2008;3:70–8.
Watarai H, Fujii S, Yamada D, et al. Murine induced pluripotent stem cells can be derived from and differentiate into natural killer T cells. J Clin Invest. 2010;120:2610–8.
Seki T, Yuasa S, Oda M, et al. Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell. 2010;7:11–4.
Soldner F, Laganiere J, Cheng AW, et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell. 2011;146:318–31.
Chen F, Pruett-Miller SM, Huang Y, et al. High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat Methods. 2011;8:753–5.
Author information
Authors and Affiliations
Corresponding authors
About this article
Cite this article
Watarai, H., Yamada, D., Fujii, Si. et al. Induced pluripotency as a potential path towards iNKT cell-mediated cancer immunotherapy. Int J Hematol 95, 624–631 (2012). https://doi.org/10.1007/s12185-012-1091-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-012-1091-0