
Abstract
Stem cells exhibit a number of characteristic features, including the capacity for self-renewal and differentiation into multiple cell types, stress resistance, and drug efflux activity. These specific biological characteristics are supported by signals from the surrounding niche and the stemcell-specific transcription factor set, including hypoxia and the machinery that senses low oxygen levels. These properties are essential for normal stem cells, and when defective may induce cellular senescence and tumorigenesis. In contrast, cancer stem cells in tumor tissue utilize these biological characters driven by stemcell-specific molecular mechanisms and acquire indefinite self-renewal capacity, drug resistance, and metastatic ability. A fuller understanding of the differences between normal and malignant stem cells in the biological and molecular context is, therefore, necessary to the development of therapies against cancer stem cells. In this review, we discuss the effect of hypoxic microenvironment on normal and malignant stem cells and describe their molecular machinery with an emphasis on hematopoietic stem cells and their malignant counterparts, leukemic stem cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Molecular oxygen (O2) plays a key role in the production of the currency of cellular energy, adenosine triphosphate (ATP), in mitochondria. O2 is inhaled and incorporated from lung to bloodstream, and is delivered to peripheral tissues via red blood cells through binding to hemoglobin. After reaching peripheral tissue, O2 is released from hemoglobin and is passively diffused into every cell. Partial pressure of O2 in the peripheral tissue is, therefore, lower than atmospheric oxygen, and varies by cell type [1]. Interestingly, stem cells, which occupy the top tier of the organ differentiation hierarchy, often localize at hypoxic microenvironments within tissues. Stem cells have two essential abilities: self-renewal and multilineage differentiation [2, 3]. These are maintained by a microenvironment suitable for stem cells (niche) by various factors, including cytokines, growth factors, adhesion molecules, and extracellular matrix derived from surrounding niche cells [4]. The hypoxic stem cell activates various molecular machineries to respond and adapt to the hypoxic environment [5–7].
In contrast to normal tissue, most tumor tissues are supported by the nutrient vessels generated by abnormal angiogenesis. Because the tumor vasculature differs significantly from functional, mature blood vessels, the tumor is poorly perfused and there are hypoxic areas in the tumor tissue [8]. Several tumors are thought to be maintained by a subpopulation called cancer stem cells [9]. The pool size, biological character and dynamics of cancer stem cells contribute to tumor development, progression, drug resistance, and metastasis in vivo. Accordingly, cancer stem cells are important therapeutic targets. A subset of the cancer stemcell population localizes at the hypoxic niche and maintains itself by the hypoxia response molecular system [6]. In this article, we will overview the hypoxia-responsive molecular machinery and describe its involvement in the maintenance of normal and malignant hematopoietic stem cells.
Hematopoietic stem cell
The hematopoietic stem cell (HSC) is one of the best characterized tissue stemcell types. Mammalian adult HSCs are localized in bone marrow, where PO2 and oxygen saturation are relatively low compared to other organs [10]. Low O2, therefore, appears to be a niche component for HSCs. O2 generates reactive oxygen species (ROS) through mitochondrial electron transport chain or cytosolic/phagosomal NADPH oxidase. Exposure to aberrant levels of ROS induces senescence dysfunction in stem cells [11], resulting in perturbed homeostasis at the organ (and whole body) level. Clearly it is advantageous for stem cells to localize at a hypoxic niche where the ROS source O2 is limited. Recent research in stemcell biology has shed light on the precise molecular mechanism by which stem cells are maintained by the hypoxia response system.
HIF-1 as a pivot of cellular hypoxia response
HIF-1α is a bHLH-PAS-type transcriptional factor, which is essential for cellular and systemic hypoxic responses [12]. HIF-α family has three members, including HIF-1α, HIF-2α, and HIF-3α. Prolyl residues in the HIF-1α oxygen-dependent degradation domain (ODD) are hydroxylated by HIF prolyl hydroxylases (Phds) under normoxia [5, 12]. The hydroxylated ODD domain of HIF-1α is recognized by von Hippel-Lindau protein (VHL). VHL is an E3 ubiquitin ligase and is responsible for the autosomal dominant hereditary disorder von Hippel-Lindau disease, or the autosomal recessive disorder Chuvash polycythemia. If VHL is mutated, HIF-1α and HIF-2α proteins are over-stabilized by the impaired ubiquitin proteasome pathway, even under normoxic conditions. As a result, patients with von Hippel-Lindau disease or Chuvash polycythemia (mild stabilization of HIF-1α and extensive stabilization of JAK2 [13]) suffer from various symptoms due to the deregulated HIF-related pathways.
Under hypoxic conditions, Phds are enzymatically inactive and HIF-1α protein is stabilized by escaping from protein degradation [12]. Several niche factors for HSCs, such as thrombopoietin (TPO) and stemcell factor (SCF), also stabilize HIF-1α protein in hematopoietic cells even under normoxia [14, 15]. Stabilized HIF-1α forms a heterodimer with the oxygen-independent subunit HIF-β (aryl hydrocarbon receptor nuclear translocator; Arnt), translocates to the nucleus, and directly binds hypoxia response elements (HREs) in the promoter regions of HIF downstream genes, thereby activating these expressions [5, 12].
HIF system in embryonic hematopoiesis
The Arnt subunit is reportedly required for hematopoietic cell generation during ontogeny. Arnt−/− mutation results in vascular and hematopoietic defects by 10.5 dpc [16, 17]. Arnt−/− embryos exhibit increased numbers of apoptotic hematopoietic cells, defective vasculogenesis and angiogenesis. Using para-aortic splanchnopleural (P-Sp) explant cultures [18], these defects are rescued by the addition of normal Sca-1+ hematopoietic cells or VEGF protein [19]. These results suggest that HIF/Arnt heterodimer coordinates embryonic endothelial cell emergence and vessel maturation by promoting hematopoietic cell survival and paracrine VEGF production. The proliferation of embryonic multilineage hematopoietic progenitors is regulated by a hypoxia-mediated signaling pathway, as Arnt−/− embryoid bodies fail to exhibit progenitor proliferation by hypoxia [17]. Also, Arnt−/− embryos show decreased yolk sac hematopoietic progenitors. This phenotype is not cell autonomous, but rather cell extrinsic. A decreased level of in Arnt-dependent VEGF expression is the responsible mechanism. Therefore, hypoxia response system is essential for the proliferation and survival of hematopoietic progenitors during embryonic hematopoiesis. It is reported that an antagonist of aryl hydrocarbon receptor (AhR), a heterodimeric partner of Arnt, can maintain human HSCs in vitro [20]. It is interesting to see whether this is related to the HIF effect or not.
Hypoxic HSC niche in the bone marrow
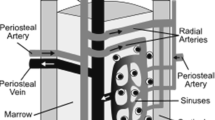
Mammalian HSCs balance cell cycle quiescence and self-renewal division, and retain the capacity for multilineage differentiation, which generates all the lineages of hematopoietic cells. After birth, HSCs localizes to the endosteal zone of the bone marrow. The endosteal zone is highly vascularized by fenestrated sinusoidal endothelium, which is easily transited by hematopoietic cells. As a result, blood flow is slow and the perfusion rate of the bone marrow cells is thought to be low. Bone marrow is densely occupied by numerous hematopoietic cells, which actively consume O2, and a simulation study suggests a 90 % reduction of PO2 at the point 100 μm away from the bone marrow vasculature [21].
Intravital time-lapse observation of the bone marrow with a multi-photon microscopy revealed that a transplanted HSC detach from bone marrow endothelium and penetrate deep into the bone marrow [22], which is thought to be hypoxic. Analysis of bone marrow hematopoietic cells following the injection of perfusion tracer into mice suggests that HSCs are poorly perfused compared to other bone marrow cells [23]. In parallel, a hypoxia probe pimonidazole is highly retained in murine HSC fraction of the bone marrow [23]. As transplanted human HSCs in immunodeficient mice reacquire the hypoxic status before they enter the quiescent steady state in cell cycle [24], the hypoxic nature of HSCs may be a common feature among species. A previous report showed that the average PO2 in the human bone marrow is 55 mmHg and oxygen saturation about 87.5 % [10]. Considering a gradual decrease of PO2 from the bone marrow vasculature, the actual PO2 at the HSC niche distant from the bone marrow vasculature is low and hypoxic. Because hypoxic culture of human HSCs elevates HIF-1α protein [25], induces quiescence [26], and supports the transplantation capacity into immunodeficient mice [25, 26], HIF-1α-mediated hypoxia response is activated and plays roles in HSC maintenance in vivo.
Regulation of adult hematopoietic stem cells by VHL/HIF regulatory system
HIF-1α deficiency is lethal to the embryo. Until embryonic day 8.5, it shows drastic morphological defects including a lack of cephalic vascularization, reduced number of somites, abnormal neural fold formation and an enhanced tissue hypoxia [27]. Therefore, the role of HIF-1α in adult HSCs must be analyzed by a conditional knockout approach using HIF-1αflox/flox mice harboring the Mx1-Cre transgene, which actuates the inducible Cre recombinase expression after synthetic double strand RNA poly I:C in various tissues including hematopoietic cells. Inducible knockout of HIF-1α by Mx1-Cre in mice (HIF-1αΔ/Δ mice) results in a loss of stress resistance in HSCs during serial bone marrow transplantation, treatment using chemotherapeutic agents, and aging [28]. These stresses induce the loss of the HSC pool in the bone marrow. After the bone marrow transplantation assay, the expression levels of Ink4a locus products, p16Ink4a and p19Arf which contribute to cellular senescence are clearly elevated in HIF-1αΔ/Δ HSCs. Because suppression of p16Ink4a and p19Arf restores the transplantation capacity of HIF-1αΔ/Δ HSC, HIF-1α protects HSCs from stress-induced cellular senescence in vivo. Loss of cell cycle quiescence in HIF-1αΔ/Δ HSCs suggests that an aberrant proliferation of HSC is a cause of senescence in HIF-1αΔ/Δ HSCs.
Heterozygous deletion of VHL (VHL+/Δ) shows enhanced cell cycle quiescence in HSCs [28]. Moreover, hematopoietic progenitor fraction, which is rapidly proliferating in the steady state, enters cell cycle quiescence. Therefore, hematopoietic progenitors normally do not express HIF-1α protein but are sensitive to over-stabilized HIF-1α-mediated quiescent induction. These observations suggest that the VHL/HIF-1α regulatory system plays a key role in the cell cycle quiescence of hematopoietic stem/progenitor cells in vivo. The maximized expression of HIF-1α protein in HSCs by homozygous deletion of VHL (VHLΔ/Δ) also induces quiescence in HSCs [28]. However, in clear contrast, VHLΔ/Δ HSCs completely lose the reconstitution ability of the bone marrow upon transplantation. These over-quiescence and defective transplantation phenotypes are reversed by the co-deletion of HIF-1α and VHL. Thus, these phenotypes in VHLΔ/Δ HSCs are HIF-1α dependent. A decreased homing capacity to bone marrow in VHLΔ/Δ hematopoietic stem/progenitor cells may explain the loss of stemcell capacity phenotype of VHLΔ/Δ HSCs. In addition to the protein level regulation of HIF-1α by VHL, transcriptional activation of HIF-1α by Meis1 transcription factor is involved in the anaerobic metabolic phenotype in hypoxic HSCs [29]. Because the moderate increment of HIF-1α by heterozygous deletion of VHL sustains the HSC pool during transplantation or aging [28], a modest induction of HIF-1α protein in HSCs by modulation of transcriptional or posttranscriptional methodology is thought to be useful for the ex vivo manipulation of HSCs without senescence.
Although the means by which HIF-1α maintains quiescence and metabolic features in HSCs remains unknown, several potential downstream candidates are reported (Fig. 1). First, Vegfa(δ/δ) mouse, a mutant mouse model with a mutated HRE in VEGF promoter region, shows a defective HSC phenotype in vivo [30]. HRE is recognized and bound by HIF-1 complex for the transcriptional activation. Decreased expression of Vegfa is observed in HSCs but not their progeny cells. Loss of hypoxia-regulated Vegfa expression in Vegfa(δ/δ) mouse increases the numbers of phenotypic HSCs. However, HSC function is clearly defective upon transplantation. Also, Miharada et al. [31] proposed a model that HIF-1α induces Cripto/GRP78 signaling for HSC maintenance. A subfraction of HSCs expressing GRP78, a heat shock protein, localizes in the hypoxic endosteal region and is quiescent in cell cycle, and shows a lower mitochondrial potential compared with GRP78− HSCs. Inhibition of GRP78 by a neutralizing antibody resulted in a reduction of GRP78+ HSCs in the endosteal area. Cripto, a ligand for GRP78, is expressed in various niche cells including osteoblasts, mesenchymal stem cells, and HSCs themselves. Cripto/GRP78 signaling in the hypoxic HSC niche regulates HSC quiescence by inducing high glycolysis activity in HSCs and maintains HSCs in hypoxia. The promoter region of Cripto gene has HRE. In HIF-1αΔ/Δ mice, decrease in GRP78+ HSCs and reduction of Cripto in the endosteal niche cells are observed. As Cripto is activated by other stimulus and HIF-1 activates various downstream regulators, it would be interesting to analyze the detailed function of HIF-1α/Cripto/GRP78 pathway in HSC and identify HIF-1α downstream regulator in HSCs.

Roles of HIF-1 in hematopoietic stem cells. Schematic representation of HIF-1 action on hematopoietic stem cells. Hypoxia or cytokine signaling by SCF or TPO enhances HIF-1-mediated transcriptional activation. These stimuli activate directly HIF-1 downstream target including VEGF or Cripto. Autocrine (hematopoietic stemcell-derived) or paracrine (osteoblast or mesenchymal stemcell-derived) Cripto binds to membrane GRP78 for maintaining quiescence. In addition, HIF-1α transcription is maintained by Meis1
Roles of HIFs in leukemic stem cells
In various patient samples from various hematological malignancies, HIF-1α activation is observed. Therefore, HIF-mediated signaling can play a pivotal role in maintenance of their stemcell fraction. There are several O2-dependent and -independent modulation mechanisms of HIF-1α protein in leukemic cells. Notably an O2-independent mechanism by isocitrate dehydrogenase (IDH) has been reported. IDH has two family members: IDH1 and IDH2. IDH catalyzes the interconversion of isocitrate and 2-oxoglutarate (2-OG) and is frequently mutated in human brain tumors and leukemia [32, 33]. IDH mutants have the neomorphic enzymatic ability to convert 2-OG to the (R)-enantiomer of 2-hydroxyglutarate ((R)-2HG). (R)-2HG, but not (S)-2HG, stimulates Phd enzymatic activity, resulting in decreased HIF protein levels [34]. Because IDH mutants block the differentiation of primitive hematopoietic stem/progenitor cells [35], these biochemical reactions are thought to promote transformation and generation of leukemic stem cells.
Although it is unclear whether the HIF-1α protein expression, therapeutic outcome, and prognosis are correlated, recent studies suggest that murine lymphoma model and human acute myeloid leukemia (AML) stem cells activate HIF-1α signal even under normoxia and maintain the tumor stemcell capacity (Fig. 2) [36]. Knockdown of HIF-1α and HIF inhibitor or overexpression of VHL suppresses the colony-forming capacities of murine lymphoma and human AML. The engraftment of these models in immunodeficient mice is also suppressed by the treatment of HIF inhibitor. In these models, the putative lymphoma stemcell fraction activates HIF-1α to suppress the negative feedback loop of Hes1 and increased the impact of Notch signals [36]. Retroviral transduction BCR-ABL in Vav-Cre-mediated HIF-1α−/− hematopoietic stem/progenitor cells also showed that the generation of chronic myeloid leukemia stem cells by BCR-ABL is defective under HIF-1α deficiency (Fig. 2) [37]. Although both the IDH-mediated destabilization [34] and the aberrant stabilization [36, 37] of HIF-1α are potentially involved in the pathogenesis of leukemia/lymphoma stem cells, therapeutic modulation of HIF-1α levels in leukemic stem cells may represent a potential target for the treatment of hematological malignancies.

Mechanisms of HIF-1-mediated maintenance of stem cell in hematological malignancy. Schematic representation of HIF-1 action on leukemic/lymphoma stem cells. Hypoxia or BCR-ABL enhances HIF-1-mediated transcriptional activation. These stimuli activate directly HIF-1 downstream target including VEGF or Glut1, or inhibits negative feedback loop of Notch1-Hes1 regulatory system to enhance Hes1-mediated transcription. As a result, leukemic stemcell generation and maintenance are promoted.
Conclusion
In this review, we have outlined the effects of hypoxia response system on hematopoietic and leukemic stem cells. Classical concept of tumor hypoxia is now connected to the hierarchical normal and leukemic stemcell system by the growing evidences. A deeper understanding of the similarities and differences of hypoxia response and oxygen metabolism between hematopoietic and leukemic stem cells may provide a novel approach to the therapeutic targeting of leukemic stem cells. Such knowledge may also contribute to new technologies for in vitro manipulation, maintenance, and expansion of normal hematopoietic stem cells.
References
Csete ME, Doyle JC. Reverse engineering of biological complexity. Science. 2002;295:1664–9.
Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–44.
Seita J, Weissman IL. Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip Rev Syst Biol Med. 2010;2:640–53.
Scadden DT. The stem-cell niche as an entity of action. Nature. 2006;441:1075–9.
Simon MC, Keith B. The role of oxygen availability in embryonic development and stem cell function. Nat Rev Mol Cell Biol. 2008;9:285–96.
Mohyeldin A, Garzon-Muvdi T, Quinones-Hinojosa A. Oxygen in stem cell biology: a critical component of the stem cell niche. Cell Stem Cell. 2010;7:150–61.
Suda T, Takubo K, Semenza GL. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell. 2011;9:298–310.
Semenza GL. Oxygen homeostasis. Wiley Interdiscip Rev Syst Biol Med. 2010;2:336–61.
Dick JE. Stem cell concepts renew cancer research. Blood. 2008;112:4793–807.
Harrison JS, Rameshwar P, Chang V, Bandari P. Oxygen saturation in the bone marrow of healthy volunteers. Blood. 2002;99:394.
Jang YY, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood. 2007;110:3056–63.
Semenza GL. Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem J. 2007;405:1–9.
Russell RC, Sufan RI, Zhou B, Heir P, Bunda S, Sybingco SS, et al. Loss of JAK2 regulation via a heterodimeric VHL-SOCS1 E3 ubiquitin ligase underlies Chuvash polycythemia. Nat Med. 2011;17:845–53.
Kirito K, Fox N, Komatsu N, Kaushansky K. Thrombopoietin enhances expression of vascular endothelial growth factor (VEGF) in primitive hematopoietic cells through induction of HIF-1alpha. Blood. 2005;105:4258–63.
Pedersen M, Lofstedt T, Sun J, Holmquist-Mengelbier L, Pahlman S, Ronnstrand L. Stem cell factor induces HIF-1alpha at normoxia in hematopoietic cells. Biochem Biophys Res Commun. 2008;377:98–103.
Maltepe E, Schmidt JV, Baunoch D, Bradfield CA, Simon MC. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nature. 1997;386:403–7.
Adelman DM, Maltepe E, Simon MC. Multilineage embryonic hematopoiesis requires hypoxic ARNT activity. Genes Dev. 1999;13:2478–83.
Takakura N, Watanabe T, Suenobu S, Yamada Y, Noda T, Ito Y, et al. A role for hematopoietic stem cells in promoting angiogenesis. Cell. 2000;102:199–209.
Ramirez-Bergeron DL, Runge A, Adelman DM, Gohil M, Simon MC. HIF-dependent hematopoietic factors regulate the development of the embryonic vasculature. Dev Cell. 2006;11:81–92.
Boitano AE, Wang J, Romeo R, Bouchez LC, Parker AE, Sutton SE, et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science. 2010;329:1345–8.
Chow DC, Wenning LA, Miller WM, Papoutsakis ET. Modeling pO(2) distributions in the bone marrow hematopoietic compartment. II. Modified Kroghian models. Biophys J. 2001;81:685–96.
Lo Celso C, Fleming HE, Wu JW, Zhao CX, Miake-Lye S, Fujisaki J, et al. Live-animal tracking of individual haematopoietic stem/progenitor cells in their niche. Nature. 2009;457:92–6.
Parmar K, Mauch P, Vergilio JA, Sackstein R, Down JD Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc Natl Acad Sci USA 2007;104:5431–6.
Shima H, Takubo K, Tago N, Iwasaki H, Arai F, Takahashi T, et al. Acquisition of G state by CD34-positive cord blood cells after bone marrow transplantation. Exp Hematol. 2010;38:1231–40.
Danet GH, Pan Y, Luongo JL, Bonnet DA, Simon MC. Expansion of human SCID-repopulating cells under hypoxic conditions. J Clin Invest. 2003;112:126–35.
Shima H, Takubo K, Iwasaki H, Yoshihara H, Gomei Y, Hosokawa K, et al. Reconstitution activity of hypoxic cultured human cord blood CD34-positive cells in NOG mice. Biochem Biophys Res Commun. 2009;378:467–72.
Ryan HE, Lo J, Johnson RS. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J. 1998;17:3005–15.
Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell. 2010;7:391–402.
Simsek T, Kocabas F, Zheng J, Deberardinis RJ, Mahmoud AI, Olson EN, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. 2010;7:380–90.
Rehn M, Olsson A, Reckzeh K, Diffner E, Carmeliet P, Landberg G, et al. Hypoxic induction of vascular endothelial growth factor regulates murine hematopoietic stem cell function in the low-oxygenic niche. Blood. 2011;118:1534–43.
Miharada K, Karlsson G, Rehn M, Rorby E, Siva K, Cammenga J, et al. Cripto regulates hematopoietic stem cells as a hypoxic-niche-related factor through cell surface receptor GRP78. Cell Stem Cell. 2011;9:330–44.
Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–66.
Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–34.
Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483:484–8.
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–67.
Wang Y, Liu Y, Malek SN, Zheng P, Liu Y. Targeting HIF1alpha eliminates cancer stem cells in hematological malignancies. Cell Stem Cell. 2011;8:399–411.
Zhang H, Li H, Xi HS, Li S. HIF1alpha is required for survival maintenance of chronic myeloid leukemia stem cells. Blood. 2012;119:2595–607.
Acknowledgments
We would like to thank Drs. Atsushi Hirao, Makoto Suematsu, Nobuhito Goda, Tomoyoshi Soga, Eiji Ikeda, and Randall S. Johnson for providing thoughtful insights and collaborations for this review. We would like to acknowledge our deep appreciation of fruitful discussions with the previous and current members of the Stem Cell Metabolism group of the Suda lab, especially Dr. Hirono Iriuchishima, Dr. Chiharu Kobayashi, Dr. June-Won Cheong, Dr. Ayako Ishizu, Dr. Hiroshi Kobayashi, and Ms. Wakako Yamada. Furthermore, we would like to thank Ms. Tomoko Muraki, Ms. Ayako Kumakubo and Ms. Takako Hirose for the preparation of this manuscript. K.T. is supported by the Global COE Program for Human Metabolomic Systems Biology and for Stem Cell Medicine of the Japan Society for Promotion of Science, and also in part by a Ministry of Education, Culture, Sports, Science and Technology (MEXT) Grant-in-Aid for Young Scientists (A) and a MEXT Grant-in-Aid for Scientific Research on Innovative Areas. T.S. was supported in part by a MEXT Grant-in-Aid for Scientific Research (A) and a MEXT Grant-in-Aid for Scientific Research on Innovative Areas.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Takubo, K., Suda, T. Roles of the hypoxia response system in hematopoietic and leukemic stem cells. Int J Hematol 95, 478–483 (2012). https://doi.org/10.1007/s12185-012-1071-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-012-1071-4