
Abstract
Aberrant methylation in promoter-associated CpG islands has been recognized as a major mechanism for tumor suppressor gene silencing in several malignancies. We determined the methylation status of nine tumor suppressor genes in 68 newly diagnosed MM patients by methylation-specific PCR. The frequency of promoter hypermethylation for individual genes was: CDH1, 50%; p16 INK4a, 42.8%; p15 INK4b, 16.2%; SHP1, 14.7%; ER and BNIP3, 13.2%; RARβ, 11.8%; DAPK 5.9%; and MGMT 0%. Overall, 79% of patients presented at least one hypermethylated gene. By univariate analysis, hypermethylation of DAPK (P < 0.001) and RARβ (P = 0.01) genes were identified as adverse prognostic features. Median OS of patients with hypermethylation in DAPK (4 months) and RARβ (34 months) was significantly lower than in patients without hypermethylation (median survival not reached), with values of P < 0.001 and P = 0.01, respectively. Our data suggest that DAPK and RARβ hypermethylation are adverse prognostic factors in MM. The relevance of these findings as poor prognosis indicators requires confirmation in a larger sample with longer follow-ups.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Multiple myeloma (MM) is an incurable plasma cell tumor that affects the bone marrow compartment, and is characterized by the production of monoclonal immunoglobulin (M protein) [1]. It accounts for more than 10% of all hematological malignancies and approximately 2% of cancer deaths [2]. In the recent years, the knowledge of the molecular and cytogenetics abnormalities involved in the origin and progression of the MM has increased considerably. Recently, two subsets of patients have been distinguished based on the cytogenetic findings: (a) the hyperdiploid variant (>46 to <76 chromosomes); and (b) the non-hyperdiploid variant, including the hypodiploid (up to 44–45 chromosomes), pseudodiploid (44/45 to 46/47 chromosomes with gains/losses compared with the normal karyotype), and the near tetraploid or hypotetraploid (75 or more chromosomes) [3, 4]. These subgroups are preferentially associated with additional cytogenetics abnormalities and distinct clinical outcome [3–7]. Additional data from gene expression profiling lead to the proposition that up-regulation of at least one gene of cyclin D family might represent a common, early oncogenic event [8], a reason why a molecular classification, based on cyclin D expression pattern and presence of IgH translocations, has been proposed [9].
While the knowledge of the genetic and cytogenetic abnormalities associated with the origin and progression of MM is becoming more apparent, the role of epigenetic modifications is still poorly understood. Aberrant methylation at promoter-associated CpG islands has been recognized as a major mechanism for silencing tumor suppressor genes in several malignancies, representing an epigenetic equivalent to mutations and deletions in carcinogenesis [10, 11]. To date, the role of aberrant promoter methylation in MM has been reported in several studies, mainly focusing on the methylation status of p16 INK4a, p15 INK4b, CDH1 (E-cadherin), SOCS1, and DAPK [12–20]. However, these studies have shown a highly variable prevalence of promoter hypermethylation within a same gene [12, 15, 17–19], as well as conflicting results between aberrant methylation and clinical characteristics and outcome [13, 14, 17, 19, 20].
These findings prompted our comprehensive analysis to determine the methylation status of nine tumor-suppressor gene promoters by methylation-specific PCR (MSP) in a group of 68 recently diagnosed MM patients. These genes were selected in view of: (a) their potential involvement in the pathogenesis of hematological diseases, and (b) their relevant involvement in several cellular pathways, such as DNA repair (MGMT), cell cycle regulation (p15 INK4b and p16 INK4a), cell–cell adherence (CDH1), apoptosis regulation (DAPK and BNIP3), hormonal response (RARβ and ER), and Jak/STAT3 signaling pathway (SHP1). In order to obtain further insights on the prognostic implications resulting from epigenetic silencing, we analyzed the relationship between methylation status, clinical characteristics, and overall survival (OS).
2 Patients and methods
2.1 Patients
Bone marrow (BM) aspirates from 68, previously untreated, MM patients and of ten healthy donors were collected at the Hospital Universitário Clementino Fraga Filho, Rio de Janeiro, Brazil. Samples were collected after informed consent was obtained in accordance with the Declaration of Helsinki. Diagnosis and staging classification of MM followed standard criteria [21, 22]. Treatment consisted of 3–4 cycles of VAD (vincristina, doxorubicin, and dexamethasone), followed by mobilization of peripheral blood hematopoietic stem cells with cyclophosphamide (4 g/m2) and G-CSF (10 μg/kg/day). After collecting a minimum of 4 × 108 CD34+ cells/Kg, patients received melphalan (200 mg/m2) and autologous hematopoietic stem cell infusion. After day +100 of transplant, patients were consolidated with thalidomide (200 mg/day) ± dexamethasone (40 mg/day for 4 days once a month), during a total of 12 months. The clinical and laboratorial characteristics of these 68 patients at diagnosis were extracted from medical charts. The characteristics analyzed were age, gender, MM subtype, Durie-Salmon and ISS staging systems, percentage of plasma cells in bone marrow, hemoglobin (Hb) level, white blood cell and platelets counts, serum calcium, serum creatinine, serum lactate dehydrogenase, albumin and serum β2 microglobulin levels (Table 1).
2.2 Genomic DNA extraction
Mononuclear cells were separated by Ficoll-Hypaque® (Sigma, St. Louis, MO, USA) density-gradient centrifugation. Cell lyses and DNA extraction were performed with DNAzol® according to the recommendations of the manufacturer (Invitrogen, Carlsbad, CA, USA).
2.3 Sodium bisulfite treatment and methylation-specific polymerase chain reaction (MSP)
Conversion of unmethylated cytosine to uracil residues by bisulfite treatment [23] was carried out by denaturing 1–3 μg of DNA with 3 M NaOH and sodium bisulfite for 16 h at 50°C, purification with the Wizard DNA Purification System (Promega, Madison, WI, USA), desulforation with 0.3 M NaOH, ethanol precipitation, and resuspension in water. Samples were stored at –20°C.
Cell lines used as controls for methylated alleles (Supplementary Table S1) were kindly provided by Dr. Marco Ladetto, University of Torino, Italy and Dr. Martino Introna, Mario Negri Institute, Milan, Italy. Cells were grown in RPMI 1640 (Life Technologies, Rockville, MD, USA) supplemented with 10% fetal bovine serum, and incubated in 5% CO2 at 37°C.
MSP assays were performed into two separate reactions with different specific primer pairs for discriminating between unmethylated (U-MSP) and methylated (M-MSP) regions. Bisulfite-treated DNA of ten bone marrow healthy donors and cell lines were used as controls for U-MSP and M-MSP, respectively, in analyses of nine suppressor tumor genes (p15 INK4b, p16 INK4a, DAPK, CDH1, SHP1, ER, MGMT, BNIP3, and RARβ). Primer sequences and annealing temperatures are listed in Table 2.
MSP reactions were carried out in a final volume of 25 μl, containing 10 ng of bisulfite-treated DNA, 0.5× PCR Buffer, 2 mM MgCl2, 0.01% (wt/vol) gelatine, 0.25 mM dNTPs, 10 pmol of each primer and 0.5 U Taq Platinum DNA polymerase (Invitrogen). Amplifications, carried out in a Flexigene PCR Thermocycler (Techne, Burlington, New York, USA), consisted of an initial denaturing step at 94°C (2 min) followed by 40 cycles at 94°C (30 s), annealing at a specific temperature (see Table 2) for 30 s, 72°C (30 s), and a final extension step at 72°C (10 min). PCR products were run in vertical electrophoresis plates at 10 V/cm in 7.5% non-denaturing polyacrylamide gels and visualized by silver staining. Results were based on two separate MSP reactions.
2.4 DNA sequencing
Unmethylated and methylated alleles of all genes were subsequently sequenced to confirm the efficiency of bisulfite treatment and the specificity of MSP reactions. PCR products were purified with the GFX TM PCR DNA and Gel Band Purification Kit H (GE Healthcare, Schenectady, NY, USA), bi-directionally sequenced following labeling with the DYEnamyc TM ET Terminator Cycle Sequencing Premix Kit H (GE Healthcare) in an ABI PRISM 377 automatic sequencer (Applied Biosystems, Foster City, CA, USA) and analyzed with the Sequence Navigator software (Applied Biosystems).
2.5 MSP sensitivity
The sensitivity of MSP assays was tested by serially diluting (10−1–10−4) methylated DNA from control cell lines with unmethylated DNA, treatment with sodium bisulfite, and subsequently amplifying them with specific primers.
2.6 Identification of chromosomal abnormalities by cIg-FISH
The presence of chromosome 13 deletion, t(4;14)(p16;q32) and t(11;14)(q23;q32) was analyzed by cIg-FISH, which combines FISH with cytoplasmic immuno-fluorescence designed to identify PCs [4, 6]. In all cases, specific commercial probes were used (Abbott Laboratories, Santa Clara, CA, USA).
2.7 Statistical analysis
The correlation between methylation status and clinical and laboratorial characteristics was assessed by Fisher’s exact test or Chi-square test (categorical variables) and Student’s t test (continuous variables). Odds ratios (OR) and 95% confidence intervals (95% CI) were calculated for binomial variables. Overall survival (OS) was estimated using the Kaplan–Meier method. Differences between survival curves were estimated by the log-rank test. Results were considered significant with P < 0.05. Data were analyzed by using SPSS software, version 10.0.
3 Results
3.1 MSP sensitivity and specificity
We analyzed CpG islands spanning along the promoter region of nine tumor suppressor genes. In all cases, the promoter hypermethylation of these genes was previously found to be associated with transcriptional silencing [14, 23–31]. The MSP specificity was confirmed by DNA sequencing, with U-MSP amplifications showing that all cytosines had been converted to uracils by bisulfite treatment, while in the M-MSP amplifications, the methylated cytosine residues in CpG dinucleotides had remained unaffected and all the unmethylated cytosines had been changed to uracils (Fig. 1). The sensitivity of M-MSP varied between 10−2 for BNIP3 and MGMT to 10−3 for p15 INK4b, p16 INK4a, CDH1, ER, DAPK, RARβ, and SHP1 (Fig. 2).

Sequencing of bisulfite-converted methylated (a) and unmethylated DNA samples (b) for p16 INK4a promoter region. The converted cytosines in the CpG dinucleotides are showed inside boxes

The sensitivity of MSP assays was tested by serially diluting (10−1–10−4) methylated DNA from control cell lines with unmethylated DNA, treatment with sodium bisulfite, and subsequently amplifying them with specific primers
To confirm the absence of non-specific reactions, we tested ten peripheral blood samples obtained from bone marrow health donors. In all the ten-health donor’s samples, the nine genes were successfully amplified with specific U-MSP primers but none of them was amplified by using M-MSP specific primers.
3.2 Methylation status in MM patients
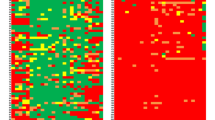
We examined the DNA methylation status of nine genes in 68, previously untreated, MM patients (Fig. 3). The frequency of hypermethylation at promoters was: CDH1, 50%; p16 INK4a, 42.6%; p15 INK4b, 16.2%; SHP1, 14.7%; ER and BNIP3, 13.2%; RARβ, 11.8%; and DAPK 5.9% (Fig. 4a). No patient showed hypermethylation at the MGMT promoter. At least one hypermethylated gene was identified in 79.4% of patients (54 out of 68). One hypermethylated gene was observed in 27.9% of patients (19/68), two genes in 25% (17/68), three genes in 17.6% (12/68), four genes in 7.3% (5/68), and five hypermethylated genes in 1.5% (1/68; Fig. 4b). The methylation profile of the nine tumor suppressor genes analyzed is shown in Fig. 5.

MSP employing specific primers for unmethylated (U) and methylated (M) DNA. In the gels is shown the methylation status in four patients for genes p15 INK4b (a), ER (b), RARβ (c), and SHP1 (d)

a Histogram showing the prevalence of the hypermethylated alleles of the different tumor suppressor genes included in this study. b Distribution of genes hypermethylated, showing the prevalence of patients with 0, 1, and more genes affected

Methylation profile of nine tumor suppressor genes in 68 MM patients. Gray boxes indicate methylated promoters, white boxes indicate unmethylated promoters
3.3 Association between aberrant methylation, clinical characteristics, and prognosis
We looked for potential correlations between methylation status and clinical parameters such as age, gender, MM subtype, Durie-Salmon and ISS staging systems, Hb level, white blood cell and platelets counts, serum calcium, serum creatinine, serum lactate dehydrogenase, albumin and serum β2 microglobulin levels.
CDH1 and p16INK4a were initially analyzed, as they were the two most commonly methylated genes. No differences in prognostic factors were observed between methylated and unmethylated groups. Durie-Salmon and ISS staging systems were also analyzed, and no correlation was found (Supplementary Table S2).
Patients with DAPK hypermethylation were more likely to have a serum creatinine level >2.0 mg/dL (P = 0.006), serum calcium >9.5 mg/dL (P = 0.05), and Durie-Salmon stage III (P = 0.04). The correlation between DAPK methylation status and clinical characteristics is shown in Table 3. Aberrant ER methylation was associated with lower albumin levels (P = 0.02), lower Hb levels (P = 0.03), high (>5.5 mg/L) serum β2-microglobulin levels (P = 0.03), and advanced ISS stages (P = 0.04). The methylation status of the other genes did not correlate with any clinical parameter.
We also search for correlations between number of methylated genes per patient and prognostic factors (Hb, platelets, serum creatinine, and β2-microglobulin levels), but there were no differences. Durie-Salmon and ISS staging systems were also analyzed, and no correlation was found with number of methylated genes (Supplementary Tables S3, S4). Of note, the patient with methylation in five genes was staged as 3 in ISS and 3b in DS system.
Likewise, we did not find any correlation between the methylation status of any gene and the presence of t(4;14)(p16;q32), chromosome 13 deletion, or t(11;14)(q23;q32) by cIg-FISH, which are associated with poor, intermediate, and good prognosis, respectively.
Univariate analysis identified hypermethylation of DAPK (P < 0.001) and RARβ (P = 0.01), platelet counts <100,000/mm3 (P < 0.001) and serum calcium >9.5 mg/dL (P = 0.03) as adverse prognostic features (Table 4), while a trend to high serum creatinine levels was not statistically significant (P = 0.064). The median OS estimates of patients with DAPK or RARβ hypermethylated (4 and 34 months, respectively) was considerably lower than in patients without aberrant methylation (median survival not reached, P < 0.001 and P = 0.01, respectively; Fig. 6). Patients with and without CDH1 and p16 INK4a had median survival of 60 months versus not reached median survival (p log rank = 0.79) and 34 versus 60 months (p log rank = 0.92), respectively.

Overall survival of 68 MM patients according to the methylation status of DAPK (a) and RARβ (b) genes. Solid line unmethylated cases; dotted lines methylated cases
4 Discussion
In recent years, aberrant methylation of 5′ promoter regions has been clearly shown to operate as a transcription regulator, and its role in several human cancers has been reported [10, 11, 32–34]. The aim of this study was to determine the methylation profile of nine tumor suppressor genes from several cellular pathways in MM patients and its association with clinical parameters and survival.
A critical point in MM genomic studies is the separation of the CD138+ cell fraction, which includes the tumor clone. However, one benefit of MSP technique is the sensitivity. Here, we calculated a sensitivity of 10−3 for the majority of genes, thus allowing the identification of methylated alleles present in at least 1% fraction. For that reason, the tumor clone isolation through sorting is not mandatory for MSP, and the technique can be performed from the mononuclear fraction.
Hypermethylation was commonly detected in >40% of cases at CDH1 and p16 INK4a promoters, and in less extent (<20%) at p15 INK4b, SHP1, ER, BNIP3, RARβ, and DAPK promoters, while it was absent at the MGMT promoter. At all, we found at least one hypermethylated gene in 79.4% of MM patients. These data suggest that aberrant methylation is a commonly observed process, affecting a variety of tumor suppressor genes controlling different pathways in MM patients.
In this study, DAPK hypermethylation showed a statistically significant association with other unfavorable clinical variables, such as higher serum creatinine levels, serum calcium, and Durie-Salmon stage III (P = 0.04). Furthermore, our data show lower median OS estimates in patients with DAPK hypermethylation than those without the aberrant methylation status (P = 0.01). Ng et al. [12] have found an association between DAPK hypermethylation and a threefold reduction in the median survival of MM patients when compared with the group without aberrant methylation, but this association was not statistically significant (P = 0.38). The DAPK protein is a serine/threonine kinase implicated in diverse apoptosis pathways, including those involved in neuronal cell death and tumor suppression [35]. Loss of DAPK expression by promoter hypermethylation has been commonly identified in a number of B-cell malignancies, especially in follicular lymphoma, MALT lymphoma, diffuse large B-cell lymphoma, mediastinal large B-cell lymphoma, and Burkitt’s lymphoma [18]. The inactivation of apoptotic pathways product of DAPK silencing may collaborate with the survival advantage conferred to lymphoma cells by BCL-2 deregulation in FL and NF-kB activation in MALT lymphoma [36].
RARβ hypermethylation was also associated with a poor outcome. This gene can mediate response to retinoid therapies, and it was suggested that lack of response might be associated to promoter hypermethylation [37, 38]. Aberrant RARβ methylation was detected with a range of very low [17] to intermediate frequency in MM patients [30], without any identifiable association between methylation status and clinical variables. In this study, we identified RARβ hypermethylation as an adverse prognostic feature by univariate analysis with the median OS in patients with RARβ hypermethylation of 34 months compared with median survival not reached in patients without abnormal methylation profile (P = 0.01).
Methylation at any other gene promoter herein investigated was not associated with differences in OS. The methylation status of p16 INK4a has been previously studied, but its biological and clinical implications in multiple myeloma are still unclear. Initial reports have shown an association between p16 INK4a hypermethylation and poor outcome [13, 14, 17]. Our results did not show a negative prognostic impact of p16 INK4a hypermethylation, according with the two larger MM cohorts previously studied, in which overall survival differences between patients with or without p16 INK4a methylated could not be demonstrated [19, 20]. It is therefore likely that p16 INK4a hypermethylation, though not associated with a poor outcome, might be a marker of disease progression.
In summary, this study showed that aberrant gene promoter methylation is a common phenomenon, affecting several metabolic pathways in MM patients. Our data suggested that DAPK hypermethylation might be an adverse prognostic factor in MM. To assess the relevance of this indicator of poor prognosis in addition to classical prognostic factors, studies with larger cohorts and longer follow-ups are required. The apparent lack of clinical impact resulting from hypermethylation of several genes herein studied suggested that their inactivation could be indicators of disease progression.
Finally, hypermethylation-associated gene silencing is a potentially reversible phenomenon, and demethylation agents have been shown to exert clinical activity in patients with myelodysplastic syndromes and acute myeloid leukemia [39, 40]. Thus, the simultaneous aberrant methylation of multiple gene promoters in MM not only provides new insights into the biology of MM but also suggests that these epigenetically affected regions may become potential targets of demethylating therapeutic strategies.
References
Kyle RA, Rajkumar SV. Multiple myeloma. N Engl J Med. 2004;351:1860–73.
Ries LAG, Eisner MP, Kosary CL. SEER cancer statistics review 1975-2001. National Cancer Institute; 2004.
Smadja NV, Fruchart C, Isnard F, Louvet C, Dutel JL, Cheron N, et al. Chromosomal analysis in multiple myeloma: cytogenetic evidence of two different diseases. Leukemia. 1998;12:960–9.
Debes-Marun C, Dewald G, Bryant S, Picken E, Santana-Dávila R, González-Paz N, et al. Chromosome abnormalities clustering and its implications for pathogenesis and prognosis in myeloma. Leukemia. 2003;17:427–36.
Fonseca R, Barlogie B, Bataille R, Bastard C, Bergsagel PL, Chesi M, et al. Genetics and cytogenetics of multiple myeloma: a workshop report. Cancer Res. 2004;64:1546–58.
Fonseca R, Debes-Marun CS, Picken EB, Dewald GW, Bryant SC, Winkler JM, et al. The recurrent IgH translocations are highly associated with nonhyperdiploid variant multiple myeloma. Blood. 2003;102:2562–7.
Smadja NV, Leroux D, Soulier J, Dumont S, Arnould C, Taviaux S, et al. Further cytogenetic characterization of multiple myeloma confirms that 14q32 translocations are a very rare event in hyperdiploid cases. Gene Chromosome Cancer. 2003;38:234–9.
Bergsagel P, Kuehl M. Critical roles for immunoglobulin translocations and cyclin D dysregulation in multiple myeloma. Immunol Rev. 2003;194:96–104.
Bergsagel PL, Kuehl M, Zhan F, Sawyer J, Barlogie B, Shaughnessy J. Cyclin D dysregulation: an early and unifying pathogenic event in multiple myeloma. Blood. 2005;106:296–303.
Laird P, Jaenisch R. DNA methylation and cancer. Hum Mol Genet. 1994;3:1487–95.
Jones P, Baylin S. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–28.
Ng M, To W, Lo KW, Chan S, Tsang KS, Cheng SH, et al. Frequent death-associated protein kinase promoter hypermethylation in multiple myeloma. Clin Cancer Res. 2001;7:1724–9.
Mateos M, García-Sanz R, López-Pérez R, Moro M, Ocio E, Hernández J, et al. Methylation is an inactivating mechanism of the p16 gene in multiple myeloma associated with high plasma cell proliferation and short survival. Br J Haematol. 2002;118:1034–40.
Guillerm G, Depil S, Wolowiec D, Quesnel B. Different prognostic values of p15INK4b and p16INK4a in multiple myeloma. Haematologica. 2003;88:476–8.
Chim CS, Kwong YL, Fung TK, Liang R. Methylation profiling in multiple myeloma. Leuk Res. 2004;28:379–85.
Chim C, Fung T, Cheung W, Liang R, Kwong Y. SOCS1 and SHP1 hypermethylation in multiple myeloma: implications for epigenetic activation of the Jak/STAT pathway. Blood. 2004;103:4630–5.
Galm O, Wilop S, Reichelt J, Jost E, Gehbauer G, Herman J, et al. DNA methylation changes in multiple myeloma. Leukemia. 2004;18:1687–92.
Rossi D, Capello D, Gloghini A, Franceschetti S, Paulli M, Bhatia K, et al. Aberrant promoter methylation of multiple genes throughout the clinico-pathologic spectrum of B-cell neoplasia. Haematologica. 2004;89:154–64.
Seidl S, Ackermann J, Kaufmann H, Keck A, Nösslinger T, Zielinski C, et al. DNA-methylation analysis identifies the E-cadherin gene as a potential marker of disease progression in patients with monoclonal gammopathies. Cancer. 2004;100:2598–606.
Gonzalez-Paz N, Chng W, McClure R, Blood E, Oken M, Van Ness B, et al. Tumor suppressor p16 methylation in multiple myeloma: biological and clinical implications. Blood. 2007;109:1228–32.
Durie B, Salmon S. A clinical staging sytem for multiple myeloma. Correlation of measured myeloma cell mass with presenting clinical features, response to treatment, and survival. Cancer. 1975;36:842–54.
The International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003;121:749–57.
Herman J, Graff J, Myohanen S, Nelkin B, Baylin S. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821–6.
Graff J, Herman J, Myohanen S, Baylin S, Vertino P. Mapping patterns of CpG island methylation in normal and neoplastic cells implicates both upstream and downstream regions in de novo methylation. J Biol Chem. 1997;272:22322–9.
Lapidus R, Nass S, Butash K, Parl F, Weitzman S, Graff J, et al. Mapping of ER gene CpG island methylation-specific polymerase chain reaction. Cancer Res. 1998;58:2515–9.
Katzenellenbogen R, Baylin S, Herman J. Hypermethylation of the DAPKinase CpG islands is a common alteration in B cell malignancies. Blood. 1999;93:4347–53.
Oka T, Ouchida M, Koyama M. Gene silencing of the tyrosine phosphatase SHP1 gene by aberrant methylation in leukemias/lymphomas. Cancer Res. 2002;62:6390–4.
Esteller M, Garcia-Foncillas J, Andion E, Goodman S, Hidalgo O, Vanaclocha V, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343:1350–4.
Esteller M, Gaidano G, Goodman S, Zagonel V, Capello D, Botto B, et al. Hypermethylation of the DNA repair gene O(6)-methylguanine DNA methyltransferase and survival of patients with diffuse large B-cell lymphoma. J Natl Cancer Inst. 2002;94:26–32.
Takahashi T, Shivapurkar N, Reddy J, Shigematsu H, Miyajima K, Suzuki M, et al. DNA methylation profiles of lymphoid and hematopoietic malignancies. Clin Cancer Res. 2004;10:2928–35.
Murai M, Toyota M, Satoh A, Suzuki H, Akino K, Mita H, et al. Aberrant DNA methylation associated with silencing BNIP3 gene expression in haematopoietic tumours. Br J Cancer. 2005;92:1165–72.
Jones P, Laird P. Cancer epigenetics comes of age. Nat Genet. 1999;21:163–7.
Baylin S, Herman J. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet. 2000;16:168–74.
Esteller M, Corn PG, Baylin SB, Herman JG. A gene hypermethylation profile of human cancer. Cancer Res. 2001;61:3225–9.
Deiss L, Feinstein E, Berissi H, Cohen O, Kimchi A. Identification of a novel serine/threonine kinase and a novel 15-kD protein as potential mediators of the gamma interferon-induced cell death. Genes Dev. 1995;9:15–30.
Sanchez-Beato M, Sanchez-Aguilera A, Piris MA. Cell cycle deregulation in B-cell lymphomas. Blood. 2003;101:1220–35.
Cote S, Momparler R. Activation of the retinoic acid receptor beta gene by 5-aza-2’-deoxycytidine in human DLD-1 colon carcinoma cells. Anticancer Drugs. 1997;8:56–61.
Virmani A, Rathi A, Zochbauer-Muller S, Sacchi N, Fukuyama Y, Bryant D, et al. Promoter methylation and silencing of the retinoic acid receptor-beta gene in lung carcinomas. J Natl Cancer Inst. 2000;92:1303–7.
Silverman LR, Holland JF, Weinberg RS, Alter BP, Davis RB, Ellison RR, et al. Effects of treatment with 5-azacytidine on the in vivo and in vitro hematopoiesis in patients with myelodysplastic syndromes. Leukemia. 1993;7(Suppl 1):21–9.
Silverman L, Demakos E, Peterson B, Kornblith AB, Holland JC, Odchimar-Reissig R, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. 2002;20:2429–40.
Acknowledgments
We are grateful to Dr. Héctor N Seuánez (INCa - UFRJ, Brazil) for critical review of this manuscript. We thank Dr. Ladetto, Alberto Rocci (University of Torino, Italy) and Dr. Introna (Mario Negri Institute, Milan, Italy) for providing the cell lines. We thank the Instituto Nacional de Câncer (INCa), Fundação Ary Frauzino (FAF), and SwissBridge Foundation for their financial support. The work was supported in part by the Conselho Nacional de Pesquisas - CNPq - Brazil and Fundação de Amparo à Pesquisa do Estado de São Paulo - FAPESP - Brazil.
Conflict of interest statement
The authors have no interests to declare.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
About this article
Cite this article
Braggio, E., Maiolino, A., Gouveia, M.E. et al. Methylation status of nine tumor suppressor genes in multiple myeloma. Int J Hematol 91, 87–96 (2010). https://doi.org/10.1007/s12185-009-0459-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-009-0459-2