
Abstract
A simple analytical method combining solvent extraction and liquid chromatography-high-resolution mass spectrometry (LC-HRMS) was developed for the analysis of pesticide residues in rice bran protein powder. Owning to the high accuracy of HRMS in determination of mass to charge ratio (m/z), a suspect screen of pesticide residues was performed by LC-HRMS prior to quantification analysis. Based on the theoretical m/z, four pesticides including isoprothiolane (IPT), tebuconazole (TBZ), propiconazole (PCZ), and tricyclazole (TCZ) were detected and further verified with their reference standards. The solvent extraction conditions were optimized according to the signal intensity of extracted ion chromatogram (XIC) in LC-HRMS. After optimization, 50% acetonitrile solution was adopted, in which the targeted pesticides could be extracted effectively (recoveries/accuracy of > 85%) with the good reproducibility (relative standard deviation (RSD) < 10.3%). Two isotope internal standards isoprothiolane-D4 (IPT-D4) and propiconazole-D5 (PCZ-D5) were applied in quantification, and the quantification results were highly consistent with those from the standard addition method. Limit of detections (LODs) and limit of quantifications (LOQs) of the method were about 0.05–0.2 and 0.2 to 1 μg kg−1, respectively, without additional purification/enrichment for these analytical targets. The developed method was applied for the analysis of five different batches of rice bran protein samples. It was found that these four pesticide residues were all below 0.02 mg kg−1, well less than the maximum residue levels (MRLs) in the latest regulations in EU and China (0.1–5 mg kg−1). Besides the rice bran protein powder, this suspect screen followed with targeted quantification approach by LC-HRMS could also be applied for other rice derivative products analysis.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Rice is one of the most important principal food consumes around the world (Kishine et al. 2008; Ohtsubo and Nakamura, 2007), and also the source of half the daily per capita of the world’s protein supply (Chandi and Sogi, 2007). Along with the rice milling, huge amount (ca. 66 million metric tons) of rice bran is produced (Fabian and Ju, 2011). However, rice bran is not consumed as food for its high fiber content and potential hull contamination. Moreover, the rice bran contains 10–16% proteins which were revealed as more nutritionally superior to other proteins especially on hypoallergenicity and anticancer activity (Helm and Burks, 1996; Shoji et al. 2001). Therefore, the rice bran is an undervalued by-product. To make better use of the rice bran, the proteins were extracted and applied as food additives, such as in infant food (Jariwalla 2001; Marcoa and Rosell, 2008). In modern rice agriculture, due to the extensive use of pesticides/fungicides to increase the quantity and quality of rice, the residues in the rice or extracted rice bran protein products raised widely concern. It has been confirmed that some fungicides are strong endocrine disruptors, which could lead to dysfunctional of some important human organs such as liver, bladder, and kidney (Briggs 1992). As their potential harmful effects on human, it is crucial to keep these pesticide residues at a safe level or maximal residual level (MRL). Accordingly, a fast and reliable analytical method was needed for the determination of pesticide residues in rice bran protein products.
For the analysis of pesticide residues in food, most of the reported methods were developed based on chromatography and mass spectrometry (MS), such as gas chromatography-mass spectrometry (GC-MS) (Cho et al. 2016; Lawal et al. 2016; Petrarca et al. 2016) and liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Hird et al. 2014; Lee et al. 2016; Stachniuk and Fornal, 2016; Zhang et al. 2016). Since there is no additional requirement on thermal stability and volatility of the analytes, the LC-MS/MS was more versatile (Malik et al. 2010). In the LC-MS/MS quantification analysis, multiple reaction monitoring (MRM) was the most recognized scan mode for its high sensitivity and selectivity in analysis of complex food samples (Danezis et al. 2016; Hird et al. 2014). In the development of MRM methods, all the reference standards of potential targets must be acquired, because the compound-specific experimental conditions such as fragment-ions and collision energies have to be optimized. However, in case of a new sample without preliminary knowledge, a lot of potential existing pesticides had to be purchased, e.g., up to over 500 (Perez-Ortega et al. 2016; Souza Tette et al. 2016), and it was blind to unknown analytes. Mostly, the number of analytes actually existed in a real sample was much less than that of purchased standards, which lead to an extra cost of time in method development and expense in analytical standards.
To narrow down the number of analytes, a quick suspect screen of the new samples was preferred. However, the low-resolution mass spectrometers (LRMS) such as triple-quadrupole mass spectrometers, which were widely used in LC-MS/MS, were incapable of performing the suspect screen analysis due to their low resolution and mass accuracy. In contrast, the high-resolution mass spectrometers (HRMS) such as time-of-flight (TOF) MS or Orbitrap-MS could provide much more qualitative information over LRMS for their high mass accuracy, gaining a wider acceptance in recent years (Gottfried and Herebian, 2013; Mou et al. 2011; Perez-Ortega et al. 2016; Senyuva et al. 2015; Zhu et al. 2016). HRMS were able to determine the ions (molecular ions or product ions) with high accuracy at a mass error of < 2 ppm, with which the suspect screen analysis could be performed based on theoretical mass to charge ratio (m/z) of the analytes (Mol et al. 2012; Romero-Gonzalez et al. 2011). So, the potentially existing targets in a sample could be found, and the further verification and quantification could be focused on a limited number of analytes. Meanwhile, as the importance and acceptance of HRMS increased and few HRMS-based methods were available for rice derivative products analysis, the development of analytical methods based on HRMS was necessary for these samples.
In this work, aiming to simplify the quantification analysis of pesticides in rice bran protein, a method for targeted suspect screen analysis was firstly developed based on solid-phase extraction (SPE) and LC-HRMS. After screen, four fungicides including isoprothiolane, tebuconazole, propiconazole, and tricyclazole were found and verified with their reference standards. With this screen analysis, the cost in time and expense on standards was greatly minimized during the quantification method development. Focused on the four screened analytes, a reliable analytical method consisting of solvent extraction and LC-HRMS was developed for the simultaneous determination of these pollutants in rice bran protein samples.
Materials and Methods
Chemicals and Reagents
Standards isoprothiolane (IPT), tebuconazole (TBZ), propiconazole (PCZ), tricyclazole (TCZ), carbendazim, bensulfuron, dimethoate, triadimefon, imidacloprid, acetochlor, and buprofezin were purchased from Macklin Biochemical Company (Shanghai, China). Isotope internal standards of isoprothiolane-D4 (IPT-D4, 100 μg mL−1 in acetone) and propiconazole-D5 (PCZ-D5, 100 μg mL−1 in acetone) were purchased from Toronto Research Chemicals Inc. (Toronto, ON, Canada) and Dr. Ehrenstorfer GmoH (Augsburg, Germany), respectively. Methanol, acetonitrile (ACN), formic acid (FA), and water for mobile phase were of LC-MS grade (Aladdin Inc., Shanghai, China). All other chemicals were of analytical grade if not stated. Rice bran protein samples were donated by a local food factory and kept in dark at room temperature.
Sample Preparation for Screen Analysis
To extract the analytes from the sample, firstly, 1.0 g rice bran protein powder was placed in a 50-mL centrifuge tube, and then 10 mL 80% ACN solution was added. After shaking by a vortex mixer for 5 min, the mixture was centrifuged at 4500 r/min for 5 min. The supernatant (5 mL) was transferred into a 10-mL centrifuge tube and evaporated to about 500 μL by nitrogen steam at 45 °C. The concentrated sample was reconstituted to 5 mL with water (resulted in a 10% ACN solution) for further treatment.
SPE was an effective method to decrease the interferences from matrix, enrich the analytes, and lower the detection limit (Grimalt and Dehouck, 2016; Pareja et al. 2011; Stachniuk et al. 2016; Wang et al. 2014). In this work, a SPE was applied to clean up the sample matrix for suspect screen analysis. HLB cartridges (Oasis HLB, Waters) were chosen for its wide adsorption capability from moderate polar to nonpolar analytes (Gilbert-Lopez et al. 2007; Perez-Fernandez et al. 2016). The washing and elution in SPE process were briefly optimized with ten widely used pesticides (IPT, TCZ, TBZ, PCZ, dimethoate, imidacloprid, acetochlor, carbendazim, bensulfuron, and buprofezin), and applied in this sample preparation. In detail, the SPE cartridge (3 mL, 60 mg) was firstly activated with 5 mL methanol and equilibrated with 5 mL H2O. After loading the reconstituted sample (5 mL), the cartridge was washed with 1 mL 20% ACN and eluted with 2 mL 80% ACN. The eluent from SPE was further evaporated by nitrogen stream at 45 °C to less than 200 μL and reconstituted to 500 μL with water. If not analyzed immediately by LC-HRMS, the pretreated samples were stored at 4 °C.
Sample Preparation for Quantification Analysis
In the quantification analysis, the solvent extraction conditions were further optimized. Firstly, 1.0 g rice bran protein powder was placed in a 10-mL centrifuge tube, and 5 mL ACN solution at different concentration (20, 50, 80, and 100%, v/v, ACN/H20) was added. After shaking by a vortex mixer for 5 min, the mixture was centrifuged at 4500 r/min for 5 min. After that, 1 mL supernatant was transferred into a 2-mL centrifuge tube and filtered by 0.22 μm membrane before LC-HRMS analysis. In contrast, SPE was also applied for cleaning up the sample. The supernatant (1 mL) was mixed with 4 mL water (total volume of 5 mL, 20% ACN) for SPE loading. The SPE and condensation conditions were the same as previous described in the “Sample Preparation for Screen Analysis” section.
LC-HRMS Analysis
The LC separation was performed on an Ultimate 3000 HPLC system (Thermo Fisher Scientific), equipped with a binary pump, an autosampler, a C18 column (Ultimate XB-C18, 3 μm, 2.1 × 150 mm, Welch Material Inc., Shanghai, China) and a column oven (kept at 40 °C). For the suspect screen analysis, the separation was firstly optimized with ten pesticides of different polarity. Mobile phase: (A) water containing 0.1% FA and (B) ACN containing 0.1% FA. Gradient: 0–2 min, 30–80% B; 2–4 min, 80% B; 4–4.1 min, 80–95% B; 4.1–7 min, 95% B; 7–7.1 min, 95–30% B; 7.1–8 min, 30% B. Flow rate, 0.4 mL min−1; injection volume, 20 μL. The total LC separation for one injection was achieved in less than 9 min, including sample loading, column cleaning, and equilibration.
The LC system was coupled to a Q-Exactive Orbitrap MS (Thermo Fisher Scientific) with an electrospray ionization (ESI) source. In the suspect screen analysis, both positive and negative full scan modes were performed in two separated LC-HRMS runs. The scan m/z range was set from 100 to 800 because the molecular weights of most pesticides were in this range (Cotton et al. 2016). The ESI parameters: sheath gas flow rate, 40 psi; aux gas flow rate, 10 psi; spray voltage, 4 kV; capillary temperature, 320 °C; S-lens level, 55 and aux gas heater temperature, 350 °C. The MS resolution of 140,000 FWHM (full width at half maximum) and 70,000 FWHM were applied in the screen analysis and quantification analysis, respectively. Data were acquired and analyzed by Xcalibur 3.0 (Thermo Fisher Scientific). For the extraction of chromatograms (extracted ion chromatogram, XIC) in suspect screen, a mass error of 2 ppm (mass tolerance window) was applied, which could provide adequate mass accuracy for qualification (Acena et al. 2015). And mass error of 5 ppm was applied for the quantification analysis since specificity of LC-HRMS at such accuracy was comparable or superior to that of LC-MS/MS with LRMS (Farre et al. 2014). After screen analysis, the suspect targets were verified by their corresponding reference standards.
Quantitative Analysis
In the LC-MS analysis of a real sample, the unknown interferences would unpredictably enhance or suppress the signal intensity of the analytical targets, namely matrix effect, which could greatly affect the accuracy of the quantification results. To correct such error from the matrix effect, isotope dilution method was considered one of the most reliable methods, in which a fixed ionization efficiency ratio of targeted analyte to isotopic internal standard (IS) (analyte/IS) could be achieved (Asam and Rychlik, 2015; Mayr and Schieberle, 2012). In this work, two isotopic ISs IPT-D4 and PCZ-D5 were used, while other ISs were not available at the moment during method development. With these two isotope ISs, the matrix effect for IPT and PCZ could be effectively corrected. But for TBZ and TCZ, since the isotopic ISs (IPT-D4 and PCZ-D5) were not co-eluted and the ionization efficiency ratio of targeted analyte to isotopic IS might change among different samples. Though there were quite a few reported methods by using one isotope IS for quantification of several analytes (Andradel et al. 2017; Kim et al. 2015; Xiong et al. 2012), further verification was needed for this quantification manner (Hewavitharana et al. 2014). In this work, to minimize the random error from matrix effect in the quantification of TBZ and TCZ, two sets of quantification data were collected and averaged based on each IS (IPT-D4 and PCZ-D5). The quantification linear dynamic ranges of each analyte in solvent were determined by plotting the peak area ratio versus the concentration ratio of analyte to IS. The mixed analytes with different concentration ranging from 0.1 to 30 μg L−1 were spiked with these two ISs at 5 μg L−1 and analyzed. It showed great linearity in the tested concentration range with R2 > 0.99 (Fig. S2).
Since no blank control was available for the preparation of spiked samples, the validity of using ISs in quantification was evaluated by comparing the results with that from the standard additional method, which was also a reliable quantification method but much more laborious. In detail, for one sample, a set of five test tubes contained 1 g rice bran protein were prepared. The samples in the tubes were spiked with mixed standards at different final concentrations of 0, 1, 2, 5, and 10 μg kg−1 and analyzed at the same procedure as previous described. Finally, the calibration curves were established by plotting the peak areas against the spiked concentrations, and the intercept at x-axis was the concentration of analytes in the original samples.
Results and Discussion
Development of the LC-HRMS Method
The LC separation gradient was optimized based on the separation of ten pesticides, and all the analytes could be well separated in 8 min with good peak shape in LC-HRMS (XICs are shown in Fig. 1). Since the noise of the baseline was zero in the XIC at mass error/tolerance < 5 ppm, so the limit of detection (LOD) of the LC-HRMS was estimated as the concentration which generates a peak intensity of 1 × 104 counts (Farre et al. 2014). The LODs and LOQs of LC-HRMS for all the tested pesticides are estimated in the range of 0.02–0.1 and 0.1–1 μg L−1, respectively. All the samples were analyzed by this LC-HRMS method in the following work.

Separation of ten pesticides in LC-HRMS at the concentration of 0.1 μg g−1 (except imidacloprid and acetochlor of 1 μg g−1)
Suspect Screen Analysis
After LC-HRMS of the prepared sample, the XIC was extracted from the total ion chromatogram with a criterion of mass error < 2 ppm, intensity of isotope peak of 13C (compared with theoretical intensity) and at least 5 data points for each peak. A full list of 38 pesticides (including herbicides, fungicides) with their theoretical m/z for data process was shown in Table S1. With the assistance of software Xcalibur 3.0, four pesticide residues including IPT, TCZ, PCZ, and TCZ were found with considerably high peak intensity of > 5 × 105 (Fig. 2), and the mass error of m/z between the theoretical and experimental were all less than 1.5 ppm (Table 1). These suspected residues were further verified by their corresponding standards in LC-HRMS, and the quantification method was developed focusing on these four compounds. It was a little surprising that residue of atrazine was not detected, which was one of the most used herbicides in China (also around the world) and showed high occurrence in environmental water and soil (Mudhoo and Garg, 2011; Udikovic-Kolic et al. 2012). To further confirm this, fortified samples were prepared by spiking of atrazine to the samples at final concentrations of 1 and 10 μg L−1, and analyzed at the same conditions. The results showed that the atrazine in fortified samples could be well detected (Fig. S3), and confirmed the absence of atrazine (below the LOD) in original samples.

The XIC and theoretical m/z of the four pesticides [M+H]+ detected in the rice bran protein powder samples
For some strong polar pesticides such as glyphosate, they could not be well captured in sample loading for their weak retention, which led to poor recovery. So, the screen analysis in SPE-LC-HRMS may fail to detect these strong polar compounds. To avoid such false negative results, the screen analysis was also performed without SPE to further check the absence (below LOD) of an analyte.
Optimization of Solvent Extraction
Focusing on the screened four pesticides, different ACN contents in the extraction solvent (20, 50, 80, and 100%, v/v, ACN/H2O) were compared based on the XIC signal intensity in the LC-HRMS (mass error < 5 ppm). Since the noise of the baseline was zero, the performances of these solvents were directly compared based on the signal intensity. It was found that the extracts from 50 and 80% ACN showed overall higher signal intensity than the rest, while the performance of 50% ACN was comparable to that of 80% ACN (Fig. 3). Considering more impurities could also be extracted at high ACN concentration (80%), which would lead to more serious matrix effect, the 50% ACN was preferred for sample extraction.

Comparison of the signal response of the analytes with different extraction solvents
The performance of SPE treatment in quantification analysis was also evaluated under the previously optimized washing and elution conditions. The XIC of these four pesticides with and without SPE were compared. Owning to the elimination of interference by SPE, the signal intensity of IPT and PCZ was improved about 50 and 20%, respectively (Fig. 4). However, about 30% decrease was found for TCZ, which probably own to the sample loss in SPE for its poor hydrophobic retention. Overall, it could be seen that the pretreatment with SPE contributed a limited improvement in LODs. Considering the cost of in time, labor and expense in SPE, a simple solvent extraction without SPE with was adopted for the rice bran protein analysis.

Comparison of the signal response between with and without SPE
Verification of the Quantification Method
In LC-MS quantification, stable isotope dilution method was considered the most robust and reliable, which could greatly compensate errors from matrix effect and save analytical time (Asam et al. 2015; Wagner et al. 2005). Therefore, isotope dilution method was adopted for quantification. In this work, IPT and PCZ could be reliably quantified by their corresponding isotopic IS of IPT-D4 and PCZ-D5 (XICs of the analytical standards were shown in Fig. S1). For the quantification of TBZ and TCZ, the concentration was firstly calculated using these two isotope ISs individually, and then averaged. Usually, the co-elution in LC of an IS with the analytical targets was preferred since the same matrix at the retention time was same and the signal response ratio of the target to IS was similar. But in this work, the retention times of IPT-D4 and PCZ-D5 were different to that of TBZ and TCZ. Considering the method was developed only for one type of samples, the matrix were similar among different samples and the signal response ratio of the target to IS could be maintained. So, such quantification method was reasonable providing an acceptable accuracy was verified by other reliable methods. The accuracy of using IPT-D4 and PCZ-D5 to quantify TBZ and TCZ were further validated by comparing the results with that from standard addition method. And the results indicated these two quantification methods showed good consistency with each other, with the maximal difference at less than 20% (Table 2). Also, compared with the quantification results between each IS (for TBZ and TCZ), the random error could be greatly compromised after averaging process.
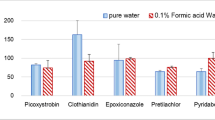
To verify accuracy, since there was no blank control, standards with known concentrations (5 and 20 μg kg−1) were directly spiked to the rice bran protein samples. In data process, the peak areas of the analytes from the spiked samples were subtracted by the un-spiked ones. As shown in Table 3, good accuracy (85–105%, n = 3) and satisfying reproducibility (RSD < 10%, n = 3) were achieved for these spiked samples. The matrix effect was also evaluated by comparing the ion intensity from spiked samples with that from pure water, and the ion suppression for these analytes was found at a range of 11–66% (Table S2). It should be noted that the recovery of solvent extraction and ion suppression in matrix effect were not involved in the data process. The LOQs of the whole analytical method (including solvent extraction) were estimated in the range of 0.1–1 μg kg−1 for these analytes. Considering the MRLs for most regulated pesticide residues were higher than 20 μg kg−1, this method was sufficient in LOQs for the analysis of food samples. These results confirmed that this method was reliable and sensitive for the quantification analysis of pesticides in rice bran protein. However, if this method would be applied in other rice derivative products analysis, an additional verification of the accuracy was needed since the matrix were different to the rice bran protein.
Analysis of Rice Bran Protein Powder Samples
With this developed methods, five rice bran protein powder samples produced in different batches (different release date) were analyzed and the results are shown in Table 4. The concentrations of these four pesticide residuals were all in the range of undetectable to 20.23 μg kg−1. According to the least regulations, the MRLs in rice and rice derivatives were 5.0, 2.0, 1.0, and 1.0 mg kg−1 for IPT, TBZ, PCZ, and TCZ in EU legislations (2011) and 1, 0.1, and 2 mg kg−1 for IPT, PCZ, and TCZ in China (Chinese National Food Safety Standard GB 2763-2014, TBZ was not regulated), respectively. Therefore, the residual levels of these four pesticides in the tasted five batches of rice bran protein products were much lower than the MRL regulated by EU and China.
Conclusion
The development of a LC-MS/MS method based on LRMS often requires high cost in purchasing of large number of standards for potential existing analytes and lots of time in optimization of compound specific MS conditions. By taking the advantage of high accuracy of HRMS in determination of m/z, a targeted suspect screen could be carried out which could greatly narrow down the number of analytes and save the expense in reference standards. In this work of analysis pesticide residues in rice bran protein, a targeted screen method was firstly developed based on SPE and LC-HRMS. Such approach could greatly cut the cost in analytical standards, and also save the time in method development since no compound specific MS conditions was needed. If more analytes beyond the suspect screen list (Table S1) needed to be considered, a retrospective analysis could be performed on the collected LC-HRMS data. In quantification analysis, while no isotope IS was available for the targeted analytes, the use of two (or more) different ISs could improve the reliability. In this quantification method, good accuracy (85–105%) and reproducibility (RSD < 10%) were achieved. With this method, five batches of rice bran protein samples were analyzed, and the results indicated that the pesticide residues in these tested samples were all well under the allowed level (1–5 mg kg−1). Furthermore, the developed analytical method without SPE reached the LOQs of 0.2–1 μg kg−1, which provided adequate sensitivity in analyzing of pesticides in rice bran protein samples. After a minor modification, this approach of suspect screen prior to quantification analysis could also be applied to other rice derivative products.
References
Acena J, Stampachiacchiere S, Perez S, Barcelo D (2015) Advances in liquid chromatography-high-resolution mass spectrometry for quantitative and qualitative environmental analysis. Anal Bioanal Chem 407(21):6289–6299
Andradel PD, Dantas RR, de Moura-Alves TLD, Caldas ED (2017) Determination of multi-mycotoxins in cereals and of total fumonisins in maize products using isotope labeled internal standard and liquid chromatography/tandem mass spectrometry with positive ionization. J Chromatogr A 1490:138–147
Asam S, Rychlik M (2015) Recent developments in stable isotope dilution assays in mycotoxin analysis with special regard to Alternaria toxins. Anal Bioanal Chem 407(25):7563–7577
Briggs, S. A. (1992). Basic guide to pesticides: their characteristics and hazards. Taylor & Francis
Chandi GK, Sogi DS (2007) Functional properties of rice bran protein concentrates. J Food Eng 79(2):592–597
Cho J, Lee J, Lim C-U, Ahn J (2016) Quantification of pesticides in food crops using QuEChERS approaches and GC-MS/MS. Food Add Contam A 33(12):1803–1816
Cotton J, Leroux F, Broudin S, Poirel M, Corman B, Junot C, Ducruix C (2016) Development and validation of a multiresidue method for the analysis of more than 500 pesticides and drugs in water based on on-line and liquid chromatography coupled to high resolution mass spectrometry. Water Res 104:20–27
Danezis GP, Anagnostopoulos CJ, Liapis K, Koupparis MA (2016) Multi-residue analysis of pesticides, plant hormones, veterinary drugs and mycotoxins using HILIC chromatography - MS/MS in various food matrices. Anal Chim Acta 942:121–138
Fabian C, Ju Y-H (2011) A review on rice bran protein: its properties and extraction methods. Crit Rev Food Sci Nutr 51(9):816–827
Farre M, Pico Y, Barcelo D (2014) Application of ultra-high pressure liquid chromatography linear ion-trap orbitrap to qualitative and quantitative assessment of pesticide residues. J Chromatogr A 1328:66–79
Gilbert-Lopez B, Garcia-Reyes JF, Mezcua M, Molina-Diaz A, Fernandez-Alba AR (2007) Determination of postharvest fungicides in fruit juices by solid-phase extraction followed by liquid chromatography electrospray time-of-flight mass spectrometry. J Agric Food Chem 55(26):10548–10556
Gottfried S, Herebian D (2013) Determination of mycotoxins in food matrices using LC-MS/MS compared with high-resolution Orbitrap (TM) MS technology. Curr Anal Chem 9(1):99–107
Grimalt S, Dehouck P (2016) Review of analytical methods for the determination, of pesticide residues in grapes. J Chromatogr A 1433:1–23
Helm RM, Burks AW (1996) Hypoallergenicity of rice protein. Cereal Foods World 41(11):839–843
Hewavitharana AK, Tan SK, Shaw PN (2014) Strategies for the detection and elimination of matrix effects in quantitative LC-MS analysis. LC GC North Am 32(1):54–64
Hird SJ, Lau BPY, Schuhmacher R, Krska R (2014) Liquid chromatography-mass spectrometry for the determination of chemical contaminants in food. Trac-Trend Anal Chem 59:59–72
Jariwalla RJ (2001) Rice-bran products: phytonutrients with potential applications in preventive and clinical medicine. Drug Exp Clin Res 27(1):17–26
Kim D, Kim B, Hyung SW, Lee CH, Kim J (2015) An optimized method for the accurate determination of nitrofurans in chicken meat using isotope dilution-liquid chromatography/mass spectrometry. J Food Compos Anal 40:24–31
Kishine M, Suzuki K, Nakamura S, Ohtsubo K (2008) Grain qualities and their genetic derivation of 7 new rice for Africa (NERICA) varieties. J Agr Food Chem 56(12):4605–4610
Lawal A, Tan GH, Alsharif AMA (2016) Recent advances in analysis of pesticides in food and drink samples using LPME techniques coupled to GC-MS and LC-MS: A review. J AOAC Int 99(6):1383–1394
Lee Y-J, Rahman MM, Abd El-Aty AM, Choi J-H, Chung HS, Kim S-W, Abdel-Aty AM, Shin H-C, Shim J-H (2016) Detection of three herbicide, and one metabolite, residues in brown rice and rice straw using various versions of the QuEChERS method and liquid chromatography-tandem mass spectrometry. Food Chem 210:442–450
Malik AK, Blasco C, Pico Y (2010) Liquid chromatography-mass spectrometry in food safety. J Chromatogr A 1217(25):4018–4040
Marcoa C, Rosell CM (2008) Effect of different protein isolates and transglutaminase on rice flour properties. J Food Eng 84(1):132–139
Mayr CM, Schieberle P (2012) Development of stable isotope dilution assays for the simultaneous quantitation of biogenic amines and polyamines in foods by LC-MS/MS. J Agric Food Chem 60(12):3026–3032
Mol HGJ, Zomer P, de Koning M (2012) Qualitative aspects and validation of a screening method for pesticides in vegetables and fruits based on liquid chromatography coupled to full scan high resolution (Orbitrap) mass spectrometry. Anal Bioanal Chem 403(10):2891–2908
Mou RX, Chen MX, Cao ZY, Zhu ZW (2011) Simultaneous determination of triazine herbicides in rice by high-performance liquid chromatography coupled with high resolution and high mass accuracy hybrid linear ion trap-orbitrap mass spectrometry. Anal Chim Acta 706(1):149–156
Mudhoo A, Garg VK (2011) Sorption, transport and transformation of atrazine in soils, minerals and composts: A review. Pedosphere 21(1):11–25
Ohtsubo K, Nakamura S (2007) Cultivar identification of rice (Oryza sativa L.) by polymerase chain reaction method and its application to processed rice products. J Agric Food Chem 55(4):1501–1509
Pareja L, Fernandez-Alba AR, Cesio V, Heinzen H (2011) Analytical methods for pesticide residues in rice. Trac-Trend Anal Chem 30(2):270–291
Perez-Fernandez V, Gentili A, Martinelli A, Caretti F, Curini R (2016) Evaluation of oxidized buckypaper as material for the solid phase extraction of cobalamins from milk: Its efficacy as individual and support sorbent of a hydrophilic-lipophilic balance copolymer. J Chromatogr A 1428:255–266
Perez-Ortega P, Lara-Ortega FJ, Garcia-Reyes JF, Gilbert-Lopez B, Trojanowicz M, Molina-Diaz A (2016) A feasibility study of UHPLC-HRMS accurate-mass screening methods for multiclass testing of organic contaminants in food. Talanta 160:704–712
Petrarca MH, Fernandes JO, Godoy HT, Cunha SC (2016) Multiclass pesticide analysis in fruit-based baby food: a comparative study of sample preparation techniques previous to gas chromatography-mass spectrometry. Food Chem 212:528–536
Romero-Gonzalez R, Aguilera-Luiz MM, Plaza-Bolanos P, Garrido Frenich A, Martinez Vidal JL (2011) Food contaminant analysis at high resolution mass spectrometry: application for the determination of veterinary drugs in milk. J Chromatogr A 1218(52):9353–9365
Senyuva HZ, Gokmen V, Sarikaya EA (2015) Future perspectives in Orbitrap (TM)-high-resolution mass spectrometry in food analysis: a review. Food Add Contam A 32(10):1568–1606
Shoji Y, Mita T, Isemura M, Mega T, Hase S, Isemura S, Aoyagi Y (2001) A fibronectin-binding protein from rice bran with cell adhesion activity for animal tumor cells. Biosci Biotechnol Biochem 65(5):1181–1186
Souza Tette PA, da Silva Oliveira FA, Correa Pereira EN, Silva G, de Abreu Gloria MB, Fernandes C (2016) Multiclass method for pesticides quantification in honey by means of modified QuEChERS and UHPLC-MS/MS. Food Chem 211:130–139
Stachniuk A, Fornal E (2016) Liquid chromatography-mass spectrometry in the analysis of pesticide residues in food. Food Anal Methods 9(6):1654–1665
Udikovic-Kolic N, Scott C, Martin-Laurent F (2012) Evolution of atrazine-degrading capabilities in the environment. Appl Microbiol Biotechnol 96(5):1175–1189
Wagner KA, Finkel NH, Fossett JE, Gillman IG (2005) Development of a quantitative method for the analysis of tobacco-specific nitrosamines in mainstream cigarette smoke using isotope dilution liquid chromatography/electrospray ionization tandem mass spectrometry. Anal Chem 77(4):1001–1006
Wang L, Zang X, Chang Q, Zhang G, Wang C, Wang Z (2014) Determination of triazole fungicides in vegetable samples by magnetic solid-phase extraction with graphene-coated magnetic nanocomposite as adsorbent followed by gas chromatography-mass spectrometry detection. Food Anal Methods 7(2):318–325
Xiong YP, Zhao YY, Goruk S, Oilund K, Field CJ, Jacobs RL, Curtis JM (2012) Validation of an LC-MS/MS method for the quantification of choline-related compounds and phospholipids in foods and tissues. J Chromatogr B 911:170–179
Zhang ZW, Li XW, Ding SY, Jiang HY, Shen JZ, Xia X (2016) Multiresidue analysis of sulfonamides, quinolones, and tetracyclines in animal tissues by ultra-high performance liquid chromatography-tandem mass spectrometry. Food Chem 204:252–262
Zhu L, Yang ST, Li GK, Zhang XG, Yang J, Lai XF, Yang GW (2016) Simultaneous analysis of tocopherols, tocotrienols, phospholipids, gamma-oryzanols and beta-carotene in rice by ultra-high performance liquid chromatography coupled to a linear ion trap-orbitrap mass spectrometer. Anal Methods 8(28):5628–5637
Acknowledgements
Financial support from the National Natural Sciences Foundation of China (Nos. 21205058, 21607020), and starting funding from Dalian University of Technology (Nos. DUT15RC(3)048, DUT17LK40) are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Minghuo Wu declares that he has no conflict of interest. Min Liu declares that she has no conflict of interest. Fumei Deng declares that she has no conflict of interest. Yufeng Hu declares that she has no conflict of interest. Lihui Sun declares that she has no conflict of interest. Jingjing Zhan declares that he has no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Not applicable.
Electronic Supplementary Material
ESM 1
(DOCX 273 kb)
Rights and permissions
About this article

Cite this article
Wu, M., Liu, M., Deng, F. et al. Simple Solvent Extraction Coupled with Liquid Chromatography-High-Resolution Mass Spectrometry for the Analysis of Pesticide Residues in Rice Bran Protein Powder. Food Anal. Methods 11, 2368–2375 (2018). https://doi.org/10.1007/s12161-018-1221-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-018-1221-2