
Abstract
A rapid and effective analytical method for determination of pydiflumetofen residues in some foods of plant and animal origin (grapes, tomatoes, wheat, pork, milk, and eggs) was developed using a modified QuEChERS (quick, easy, cheap, effective, rugged, and safe) sample preparation procedure followed by ultra-performance liquid chromatography coupled with tandem mass spectrometry (UPLC–MS/MS). Acetonitrile was served as the extraction solvent, and an octadecylsilane-dispersive solid-phase extraction (C18-dSPE) was used to cleanup the analyte, and then detected by UPLC–MS/MS. Pydiflumetofen was eluted within 3.0 min from the HSS T3 chromatography column connected to an electrospray ionization source in positive mode. The linearity of the method was excellent (R2 ≥ 0.992) in the pydiflumetofen concentration range of 10–1000 μg kg−1. The recoveries of spiked pydiflumetofen (10, 100, and 1000 μg kg−1) from the matrices were satisfactory, being between 72.0 and 110.3%, and all with relative standard deviation values of < 15.1%. The limit of quantification for pydiflumetofen was 10 μg kg−1. This study provides a method for the routine monitoring of pydiflumetofen.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Pesticides have been used to great effect to control plant diseases and hence are essential for agriculture. However, the use of chemical pesticides violates good agricultural practice can lead to pesticide residues in treated plants that are consumed directly by humans and animals (MacLachlan and Bhula 2008). What is more, pesticides tend to concentrate in fatty tissues so that they may be found in food products of animal origin such as meat, eggs, and milk (Castillo et al. 2012). Thus, the presence of these compounds in foods of animal origin may cause secondary toxicities through the food chain, which pose a potential hazard for non-target organisms including humans. The potential hazards poses to human health, including change the functions of the endocrine systems, nervous systems, and reproductive systems, have aroused public concern worldwide (Castillo et al. 2012). Consequently, it is highly desirable to determine the levels of pesticide residues in animal and plant food products to help evaluate their impact on human health.
Pydiflumetofen [3-(difluoromethyl)-N-methyl-N-[1-methyl-2-(2,4,6-trichlorophenyl)ethyl]-1H-pyrazole-4-carboxamide] (Fig. 1) is an N-methoxy-(phenyl-ethyl)-pyrazole-carboxamide-type compound that was developed by Syngenta in 2016. It is a succinate dehydrogenase inhibitor that interferes with the respiration of plant fungal pathogens by inhibiting the activities of their complex-II enzymes and thus can effectively control many plant fungus-related diseases (Avenot and Michailides 2010). This new fungicide offers a significant improvement over previously developed fungicides for control of plant pathogens such as Botrytis cinerea, Sclerotinia sclerotiorum, and Corynespora cassiicola. Pydiflumetofen has also been shown to be active against Fusarium asiaticum, which usually attacks wheat (Hou et al. 2017). Recently, pydiflumetofen was registered in Argentina as a pesticide that could be used against soybean pathogens, but the trend for pydiflumetofen is to be registered globally for use against diseases of fruits, vegetables, and other cereals, suggesting that its residue may contaminate straw ingested by animals such as swine, cattle, and chickens, and more generally, pollute foods originating from animals. It is therefore of utmost importance to monitor the levels of pydiflumetofen remaining on foods to ensure the food safety. To the best of our knowledge, however, no analytical method has been developed to determine pydiflumetofen levels. Therefore, a reliable and robust analytical method is required to detect pydiflumetofen in foods of plant and animal origin.

Chemical structure of pydiflumetofen
Analysis of pesticide residues involves two steps: sample preparation followed by chromatographic separation and determination(Wu et al. 2014). The QuEChERS (quick, easy, cheap, effective, rugged, and safe) method, introduced by Anastassiades and colleagues (Anastassiades et al. 2003), is an excellent extracting approach for pesticide residue. This method involves acetonitrile extraction accompanied by simultaneous liquid–liquid partitioning and is followed by a dispersive solid-phase extraction (dSPE). And it has already received worldwide acceptance owing to its simplicity and high throughput (Cheng et al. 2014; Dong et al. 2012; Zhang et al. 2013). In this study, the samples of plant and animal origin may contain complex matrix components including pigments, proteins, and lipophilic inclusions, which may be easily co-extracted with the target analyte (Li et al. 2013; Liu et al. 2017). Therefore, reduction of the amounts of non-volatile and semi-volatile co-extracts is a particularly challenging task. Mostly, a primary secondary amine (PSA) sorbent was used as the dSPE sorbent to bind fatty acid compounds and anhydrous magnesium sulfate was used to remove the water. Some works have produced good results with the modified QuEChERS cleanup steps with graphitized carbon black (GCB), Florisil, or C18 (Wang et al. 2015). Besides, it is reported that multi-walled carbon nanotubes (MWCNTs) function to strongly adsorb impurities for they are hollow graphene cylinders which could be used as alternative dSPE materials to PSA or other sorbents with the original QuEChERS preparation method for the further cleanup of foods by removing matrix components such as sugars and fatty acids (Zhao et al. 2012), and this method meets the requirements for pesticide analysis.
The focus of the present research is the development and validation of a rapid analytical method for the determination of pydiflumetofen in foods of plant and animal origin by using QuEChERS procedure followed by UPLC–ESI–MS/MS analysis. To achieve high recoveries and a good purification performance, different extraction solvents, different types of sorbents including traditional ones (i.e., PSA, C18, Florisil, and GCB), and the newly introduced MWCNTs of various diameters were investigated in this article. As far as we know, this is the first report establishing a simple analytical method to determine pydiflumetofen residue in plant and animal samples.
Materials and Methods
Reagents and Materials
Pydiflumetofen (98.2% purity) was provided by Syngenta (China) Investment Co. Ltd. Chromatography-grade acetonitrile and formic acid were purchased from Sigma-Aldrich (Steinheim, Germany). Analytical-grade acetonitrile, NaCl, and anhydrous MgSO4 for pesticide residue analysis were purchased from Beihua Fine-chemicals Co. Ltd. (Beijing, China). Ultra-pure water was prepared using a Milli-Q system (Bedford, MA, USA). Nylon syringe filters (pore size, 0.22 μm), PSA (40 μm), C18 (40 μm), GCB (40 μm), and Florisil (40 μm) were purchased from Agela Technologies Inc. (Tianjin, China). MWCNTs with average external diameters of 8–15, 10–20, 20–30, 30–50, and > 50 nm were purchased from Boyu Technologies Inc. (Beijing, China).
Standard stock solutions of pydiflumetofen (100 mg L−1) were prepared in pure acetonitrile. Standard working solutions at concentrations of 0.01, 0.05, 0.1, 0.5, and 1 mg L−1 were prepared from the stock solution by serial dilution with chromatography-grade acetonitrile. Corresponding matrix-matched standard solutions were prepared at concentrations from 0.01 to 1 mg L−1 by adding blank sample extracts (grape, tomato, wheat, pork, egg, and milk) to each serially diluted standard solution. All solutions were stored at 4 °C in the dark until use. These six blank samples were collected from a local market in Beijing and not applied and contaminated by the target pesticide. Authentic grape samples were obtained from a residual trial in field of Shandong Province of China and were treated with suspension concentrate at the 1.5-fold recommended dosage of 400 mg/L when they are about half the size of rape ones. Samples were randomly collected at intervals of 0 (2 h posttreatment), 1, 2, 5, 7, 14, 21, and 28 days after application. All samples were homogenized, divided into subsamples, and kept in the dark at − 20°C until they were analyzed.
Instrument
Chromatography was performed using a Waters ACQUITY UPLC system (Milford, MA, USA), an ACQUITY UPLC binary solvent manager, an ACQUITY UPLC sample manager, and an ACQUITY column heater equipped with an ACQUITY UPLC HSS T3 column (100 mm × 2.1 mm, 1.7-μm particle size). Solvent A of the mobile phase was chromatography-grade acetonitrile, and solvent B was 2 mM ammonium acetate aqueous solution. The flow rate was 0.3 mL min−1, and the gradient elution program was 0–1.2 min, 10–90% A; 1.2–2.6 min, 90% A; 2.6–3.0 min, 90–10% A; and 3.0–5.0 min, 10% A. The injection volume was 5 μL. Pydiflumetofen eluted within 3.0 min. The temperature of the column oven was 40°C, and the temperature of the sample vial holder was 5 °C.
A triple quadrupole mass spectrometer (Waters Corp., Milford, MA, USA) equipped with an electrospray ionization source was used to analyze pydiflumetofen. The nebulizer gas was 99.95% nitrogen, and the collision gas was 99.999% argon at a pressure of 2 × 10−5 MPa in a T-ware cell. MS/MS detection was carried out in the positive and negative ionization-switching mode, and the ionization intensity was optimized for the target analyte. Typical optimized conditions were a capillary voltage of 3.0 kV and source and desolvation temperatures of 150 and 500 °C, respectively. The cone and desolvation gas flows were 50 and 1000 L h−1, respectively. The multi-reaction monitoring mode used for detection of the target compound had a dwell time of 91 ms. All other MS parameters were optimized individually and are listed in Table 1. Masslynx software (version 4.1) was applied to acquire and analyze the data.
Sample Treatment
Grapes, tomatoes, wheat, pork, eggs, and milk were purchased from a supermarket and were not contaminated by the target compound. These samples were chopped and homogenized in an Ultra-Turrax homogenizer (IKA-Werke, Staufen, Germany) and then stored in a refrigerator at a temperature below − 20 °C until analysis. Aliquots (10 g) of the homogenized grapes, tomatoes, wheat, pork, eggs, and milk matrices were individually weighed into a 50-mL Teflon centrifuge tube, and an appropriate volume of a working standard solution was added. Each tube was vortexed for 30 s and then held for 30 min at room temperature to distribute the pesticide evenly (Anastassiades et al. 2003). Next, 5 mL of ultra-pure water (only for the wheat samples) and 10 mL of analytical-grade acetonitrile for all food samples were added to the tubes, which were capped and vortexed for 5 min. Subsequently, 2 g NaCl and 4 g MgSO4 were added to each tube, and all tubes were then vortexed for 5 min and centrifuged for 5 min at 2077×g. Next, for each sample, 1.5 mL of its top layer was transferred to a 2-mL single-use centrifuge tube containing 50 mg of C18 and 150 mg of anhydrous MgSO4. Each extract was vortexed for 1 min and then centrifuged for 5 min at 2077×g.
In this process, three different extraction solvents (acetonitrile, acetonitrile/0.2% (v/v) formic acid aqueous solution, acetonitrile/1% (v/v) formic acid aqueous solution) were used to assess the ability of extract pydiflumetofen from grape samples at a spiked level of 100 μg kg−1.
And the comparison of different sorbents was conducted using 50 mg of PSA, C18, Florisil, GCB, and 10 mg of MWCNTs (8–15, 10–20, 20–30, 30–50, and > 50 nm) at the same concentration level (100 μg kg−1). Then, the samples were again vortexed for 1 min and centrifuged for 5 min at RCF 207 7×g. The resulting supernatants were individually filtered through a 0.22-μm nylon syringe filter and transferred to an autosampler vial for ultra-performance liquid chromatography coupled with tandem mass spectrometry (UPLC–MS/MS).
Method Validation
The method was validated according to the following parameters: specificity, linearity, matrix effect, limit of quantification, precision, and accuracy (Niell et al. 2015). Blank samples of grape, tomato, wheat, pork, milk, and egg were analyzed to determine the specificity of the method and to search for interfering peaks occurred at the retention time that would interfere with quantification of the analyte peaks. The linearity of the method was confirmed by analyzing the solvent-based standard and the different matrix-modified standard solutions at five concentrations between 10 and 1000 μg L−1. The corresponding linear regression equations with their associated slopes, y-intercepts, and coefficients of determination are given in Table 2. The matrix effect was calculated from the slopes of the linear calibration obtained for each matrix and solvent to determine the signal suppression/enhancement. The formula used to calculate the matrix effect was matrix effect (%ME) = [(slope of the matrix-matched calibration curve − slope of the solvent calibration curve) / slope of solvent calibration curve] × 100. The limit of quantification was estimated from the smallest concentration used during recovery experiments to provide an acceptable recovery (70–120%) and a relative standard deviation (RSD) of ≤ 20%. Accuracy and precision were determined by analyzing fortified samples at three concentrations (1, 10, and 100 μg kg−1) in quintuplicate. Accuracy is expressed in terms of analytical recovery, and precision is expressed as the intra-day and inter-day RSDs (RSDr and RSDR, respectively).
Results and Discussion
Optimization of UPLC–MS/MS Parameters
We first examined and optimized the MS parameters in both positive and negative modes with the use of the 1000 μg L−1 pydiflumetofen standard solution. A greater peak intensity for the parent ion was obtained in the electrospray ionization-positive mode. Consequently, protonated molecule [M + H]+ was used as the MS/MS precursor ion, which was fragmented by direct injection at different collision energies so that the selected reaction monitoring could be optimized to achieve the greatest sensitivity. According to the European Commission Decision 2002/657/EC for an LC-MS/MS method, the identification of an analyte can be characterized according to its retention time and the relative abundance of each selected ion fragment(Wu et al. 2014). The optimized MS/MS parameters for pydiflumetofen are listed in Table 1.
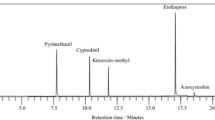
The optimum separation conditions for pydiflumetofen were investigated using 5-μL aliquots of the 1000 μg L−1 working standard solution and the UPLC HSS T3 column. The mobile phase was altered with additives because the mobile phase composition can strongly influence peak shape and the retention behavior of an analyte in an LC column, as well as the MS response (Rubert et al. 2010). To obtain a satisfactory peak shape and appropriate retention time, different mobile phase compositions (acetonitrile/water; acetonitrile/0.2% (v/v) formic acid aqueous solution; acetonitrile/2 mM ammonium acetate aqueous solution; methanol/water; methanol/0.2% (v) formic acid aqueous solution; methanol/2 mM ammonium acetate aqueous solution) were evaluated using the gradient program described in the “Materials and methods” section at 0.3 mL min−1. The greatest sensitivity and best peak shape were obtained with acetonitrile/2 mM ammonium acetate aqueous solution (Fig. 4). Using this elution system, the pydiflumetofen retention time was 2.74 min, with no interfering peaks (Figs. 2, 3, 4, and 5).

Typical UPLC–MS/MS MRM chromatograms of pydiflumetofen in a a standard sample (10 μg L−1), b a grape sample spiked at a concentration of 10 μg kg−1 pydiflumetofen, and c a blank grape sample

Effect of different sorbents (PSA, C18, GCB, Florisil, and MWCNTs) for pydiflumetofen (spiked at 100 μg kg−1) purification from different matrix (n = 5)

UPLC–MS/MS MRM chromatograms of pydiflumetofen for the effects of different mobile phase compositions. a Acetonitrile/water. b Methanol/water. c Acetonitrile/0.2% (v/v) formic acid aqueous solution. d Methanol/0.2% (v/v) formic acid aqueous solution. e Acetonitrile/2 mM ammonium acetate aqueous solution. f Methanol/2 mM ammonium acetate aqueous solution

Typical UPLC–MS/MS MRM chromatograms of pydiflumetofen in different matrix spiked at 10 μg kg−1. a A wheat sample. b A tomato sample. c An egg sample. d A milk sample. e A pork sample
Optimization of the Extraction Solvents
For analysis of pesticide residues in foods, the choice of the extraction solvent can greatly affect the extraction efficiency. Previous studies of a variety of foods have used acetonitrile as the extraction solvent for its lesser co-extracts of matrices components and satisfactory recovery (Tian et al. 2016; Zhao et al. 2015). Acetonitrile mixed with different concentrations of formic acid aqueous solution has also proved to be a good extraction solvent (Hu et al. 2015; Ju et al. 2015). We therefore assessed the ability of three different extraction solvents (acetonitrile, acetonitrile/0.2% (v/v) formic acid aqueous solution, acetonitrile/1% (v/v) formic acid aqueous solution) to extract pydiflumetofen at a spiked concentration of 50 μg kg−1 from grape samples. Although all of three extraction solvents facilitated a good recovery, as it was most convenient, acetonitrile was used as the extraction solvent in all further experiments.
Optimization of the Dispersive Solid-Phase Extraction Cleanup Process
In the cleanup procedure, the selection of the sorbent is another factor that can affect the analysis of chemical residues. After extraction of a target analyte, a purification step is usually required to remove co-extracted contaminants. Dispersive solid-phase extraction is the most commonly used purification procedure, and it uses a mixture of MgSO4 and sorbent to remove residual water and other matrix components (Wu et al. 2014). Considering the many types of chlorophyll derivatives in plant-derived foods and large amounts of proteins, lipids, and saturated and unsaturated fatty acids in animal-derived foods, the effects of the four traditional types of dispersive sorbents PSA (50 mg), C18 (50 mg), Florisil (50 mg), and GCB (20 mg) and 10 mg of each MWCNT with a different external diameter (8–15, 10–20, 20–30, 30–50, and > 50 nm) were evalued to cleanup the pydiflumetofen from the six foods (each at 100 μg kg−1). The recoveries and RSD values were satisfactory for all cleanup methods tested for all matrices (Fig. 3). Because it was the least expensive, the C18 (50 mg) treatment was selected to cleanup the samples of foods of plant and animal origin.
Method Validation
Linearity and Limit of Quantification
All calibration curves of the working standard solutions and the matrix-matched standard solutions demonstrated satisfactory linearity (Table 2). All regression coefficients for recovery of spiked pydiflumetofen from the matrices were > 0.992. The limit of quantification values were 10 μg kg−1 for all matrices.
Matrix Effects
The matrix effect, as first reported by Kebarle and Tang (Kebarle and Tang 1993), is that the presence of coextractives can affect the ionization of the target compounds by reducing or enhancing the detector response compared with that produced by the analytes in solvent. The suppression or enhancement of the response can diminish the precision and accuracy of the method. In the present study, the effects caused by the different matrices were analyzed by comparing the slopes obtained for the standards with the matrix-matched standards. In this way, no matrix effect is observed for a value of 0, values of < 0 indicate signal suppression, and values of > 0 indicate ionization enhancement. The data in Table 2 show that pronounced signal suppression was observed for pydiflumetofen in the six matrices ranged from − 35.5 to − 82.2%. The incomplete removal of matrix components may be the cause of the signal suppression. However, the underlying mechanisms by which the matrices influence the signal strength of a target compound are not thoroughly understood and need further investigation. Therefore, a matrix-matched calibration curve was performed for pydiflumetofen to eliminate the corresponding matrix effect and achieve more reliable results.
Accuracy and Precision
The accuracy and precision of the method were assessed based on the spiked blank samples at three different concentrations (10, 100, and 1000 μg kg−1). The accuracy was expressed as the recovery (%) of each spiked sample. The precision was based on reproducibility during each day and between each day and was expressed as RSDr and RSDR, respectively. The RSDr (n = 5) was calculated using the standard deviation of the recovery percentage of each set of spiked samples on the same day. The RSDR (n = 15) was measured by comparing the results of spiked samples obtained on three separate days. The mean values of the recoveries and RSDr and RSDR values of pydiflumetofen from the six samples are shown in Table 3. The mean recovery value for pydiflumetofen ranged from 76.2 to 108.0% for the six matrices. The RSDr and RSDR values ranged from 1.0 to 15.1% and from 3.2 to 12.4%, respectively. These data were consistent with the EU guidelines for pesticide residue analysis, suggesting the usefulness of the method (European Commission, 2011).
Application to Field Samples
To verify the effectiveness of this modified QuEChERS-based extraction and UPLC–MS/MS analytical method, trace levels of pydiflumetofen were assessed by analyzing grape samples obtained from a residual dynamic trial in field, carried out in China. The initial deposit of pydiflumetofen residue in the grape was 2.38 mg/kg and declined to 1.00 mg/kg by day 14. The results demonstrated the applicability of the new method for the detection of the pydiflumetofen residue. The method provided a valid method that lay the foundation for risk assessment of pydiflumetofen posed to human health.
Conclusions
We present herein a new simple and time-saving analytical method based on the QuEChERS method in conjunction with UPLC–MS/MS to determine the level of the new fungicide pydiflumetofen in foods of plant and animal origin. Under the optimized conditions, pydiflumetofen eluted within 3.0 min from the UPLC column with good specificity, recovery, linearity, precision, and accuracy. Although a strong matrix effect was observed for each examined matrix, it could be compensated for with the use of matrix-matched calibration curves. Additionally, the reliability and efficacy of the method was confirmed by determining the level of the fungicide residue in grapes grown in a field in Shandong province. In summary, the method can be used for routine monitoring of pydiflumetofen residue in foods of plant and animal origin.
References
Anastassiades M, Lehotay SJ, Stajnbaher D, Schenck FJ (2003) Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J Aoac Int 86:412
Avenot HF, Michailides TJ (2010) Progress in understanding molecular mechanisms and evolution of resistance to succinate dehydrogenase inhibiting (SDHI) fungicides in phytopathogenic fungi. Crop Prot 29(7):643–651. https://doi.org/10.1016/j.cropro.2010.02.019
Castillo M, Carbonell E, González C, Miralles-Marco A (2012) Pesticide residue analysis in animal origin food: procedure proposal and evaluation for lipophilic pesticides. In: Pesticides-Recent Trends in Pesticide Residue Assay. InTech,
Cheng Y, Dong F, Liu X, Xu J, Meng W, Liu N, Chen Z, Tao Y, Zheng Y (2014) Simultaneous determination of fipronil and its major metabolites in corn and soil by ultra-performance liquid chromatography-tandem mass spectrometry. Anal Methods 6(6):1788–1795. https://doi.org/10.1039/C3AY41742E
DG SANCO European Commission’ Directorate General for Health and Consumers. (2011)Method validation and quality control procedures for pesticide residues analysis in food and feed, European Commission, Brussels - Belgium,
Dong F, Chen X, Liu X, Xu J, Li Y, Shan W, Zheng Y (2012) Simultaneous determination of five pyrazole fungicides in cereals, vegetables and fruits using liquid chromatography/tandem mass spectrometry. J Chromatogr A 1262:98–106. https://doi.org/10.1016/j.chroma.2012.08.100
Hou Y-P, Mao X-W, Wang J-X, Zhan S-W, Zhou M-G (2017) Sensitivity of Fusarium asiaticum to a novel succinate dehydrogenase inhibitor fungicide pydiflumetofen. Crop Prot 96:237–244. https://doi.org/10.1016/j.cropro.2017.02.011
Hu M, Liu X, Dong F, Xu J, Li S, Xu H, Zheng Y (2015) Determination of ametoctradin residue in fruits and vegetables by modified quick, easy, cheap, effective, rugged, and safe method using ultra-performance liquid chromatography/tandem mass spectrometry. Food Chem 175:395–400. https://doi.org/10.1016/j.foodchem.2014.11.158
Ju C, Dong F, Liu X, Wu X, Zhao H, Zheng Y, Xu J (2015) Rapid residue analysis of oxathiapiprolin and its metabolites in typical Chinese soil, water, and sediments by a modified quick, easy, cheap, effective, rugged, and safe method with ultra high performance liquid chromatography and tandem mass spectrometry. J Sep Sci 38(6):909–916. https://doi.org/10.1002/jssc.201401126
Kebarle P, Tang L (1993) From ions in solution to ions in the gas phase-the mechanism of electrospray mass spectrometry. Anal Chem 65:972A–986A
Li M, Liu X, Dong F, Xu J, Kong Z, Li Y, Zheng Y (2013) Simultaneous determination of cyflumetofen and its main metabolite residues in samples of plant and animal origin using multi-walled carbon nanotubes in dispersive solid-phase extraction and ultrahigh performance liquid chromatography–tandem mass spectrometry. J Chromatogr A 1300:95–103. https://doi.org/10.1016/j.chroma.2013.05.052
Liu H, Yao G, Liu X, Liu C, Zhan J, Liu D, Wang P, Zhou Z (2017) Approach for pesticide residue analysis for metabolite prothioconazole-desthio in animal origin food. J Agric Food Chem 65(11):2481–2487. https://doi.org/10.1021/acs.jafc.7b00062
MacLachlan DJ, Bhula R (2008) Estimating the residue transfer of pesticides in animal feedstuffs to livestock tissues, milk and eggs: a review. Aust J Exp Agric 48(5):589–598. https://doi.org/10.1071/EA07196
Niell S, Jesús F, Pérez C, Mendoza Y, Díaz R, Franco J, Cesio V, Heinzen H (2015) QuEChERS adaptability for the analysis of pesticide residues in beehive products seeking the development of an agroecosystem sustainability monitor. J Agric Food Chem 63(18):4484–4492. https://doi.org/10.1021/acs.jafc.5b00795
Rubert J, Soler C, Mañes J (2010) Optimization of matrix solid-phase dispersion method for simultaneous extraction of aflatoxins and OTA in cereals and its application to commercial samples. Talanta 82(2):567–574. https://doi.org/10.1016/j.talanta.2010.05.008
Tian C, Xu J, Dong F, Liu X, Wu X, Zhao H, Ju C, Wei D, Zheng Y (2016) Determination of Sulfoxaflor in animal origin foods using dispersive solid-phase extraction and multiplug filtration cleanup method based on multiwalled carbon nanotubes by Ultraperformance liquid chromatography/tandem mass spectrometry. J Agric Food Chem 64(12):2641–2646. https://doi.org/10.1021/acs.jafc.6b00285
Wang P, Tian F, Xu J, Dong F, Li S, Zheng Y, Liu X (2015) Determination of flumetsulam residue in 20 kinds of plant-derived foods by ultra-performance liquid chromatography coupled to tandem mass spectrometry. Anal Methods 7(14):5772–5779. https://doi.org/10.1039/C5AY00553A
Wu X, Xu J, Dong F, Liu X, Li Y, Zheng Y (2014) Simultaneous determination of oxathiapiprolin and two metabolites in fruits, vegetables and cereal using a modified quick, easy, cheap, effective, rugged, and safe method and liquid chromatography coupled to tandem mass spectrometry. J Chromatogr A 1329:30–37. https://doi.org/10.1016/j.chroma.2013.12.087
Zhang Y et al (2013) Simultaneous determination of four neonicotinoid insecticides residues in cereals, vegetables and fruits using ultra-performance liquid chromatography/tandem mass spectrometry. Anal Methods 5:1449–1455
Zhao P, Wang L, Zhou L, Zhang F, Kang S, Pan C (2012) Multi-walled carbon nanotubes as alternative reversed-dispersive solid phase extraction materials in pesticide multi-residue analysis with QuEChERS method. J Chromatogr A 1225:17–25. https://doi.org/10.1016/j.chroma.2011.12.070
Zhao H, Xu J, Dong F, Liu X, Wu Y, Wu X, Zheng Y (2015) Simultaneous determination of three herbicides in wheat, wheat straw, and soil using a quick, easy, cheap, effective, rugged, and safe method with ultra high performance liquid chromatography and tandem mass spectrometry. J Sep Sci 38(7):1164–1171. https://doi.org/10.1002/jssc.201401234
Funding
This study was financially supported by the National Natural Science Foundation of China (NSFC, 31501678, 31672057)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This is an original research article that has neither been published previously nor considered presently for publication elsewhere.
Informed Consent
All authors named in the manuscript are entitled to the authorship and have approved the final version of the submitted manuscript. This article does not contain any studies with human or animal subjects.
Conflict of Interest
Lili Rong declares that she has no conflict of interest. Xiaohu Wu declares that he has no conflict of interest. Jun Xu declares that he has no conflict of interest. Fengshou Dong declares that he has no conflict of interest. Xingang Liu declares that he has no conflict of interest. Yongquan Zheng declares that he has no conflict of interest.
Rights and permissions
About this article

Cite this article
Rong, L., Wu, X., Xu, J. et al. Determination of Pydiflumetofen Residues in Some Foods of Plant and Animal Origin by QuEChERS Extraction Combined with Ultra-Performance Liquid Chromatography–Tandem Mass. Food Anal. Methods 11, 2682–2691 (2018). https://doi.org/10.1007/s12161-018-1178-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-018-1178-1